BACKGROUND: Megakaryocytic differentiation is a complicated process regulated by a series of transcription factors in a context- and stage-dependent manner. Recent studies have suggested that krüppel-like transcription factor 2 (KLF2) is involved in the control of embryonic erythroid precursor cell differentiation and maturation. However, the function and mechanism of KLF2 in regulating megakaryocytic differentiation remain unclear. METHODS: The expression patterns of krüppel-like transcription factors (KLFs) during megakaryocytic differentiation were identified from public databases. Phorbol 12-myristate 13-acetate (PMA) treatment of the myeloid-erythroid-leukemic cell lines K562 and HEL were used as cellular megakaryocytic differentiation models. A lentiviral transduction system was utilized to achieve the goal of amplifying or reducing KLF2. The expression of KLF2 was examined using real-time PCR and western blot. The impact of KLF2 on the megakaryocytic differentiation of K562 cells was examined by flow cytometry, Giemsa staining, Phalloidin staining and western blot. RNA-sequencing (RNA-seq) and chromatin immunoprecipitation-sequencing (ChIP-seq) technologies were used to identify the KLF2-regulated targets. RESULTS: KLF2 is increased in the maturation process of megakaryocytes. KLF2 overexpression accelerated the PMA-induced megakaryocytic differentiation, as reflected by an increased percentage of CD41/CD61 cells, an increased number of polyploid cells, and an elevated expression of P21 and P27. KLF2 knockdown exhibited the opposite results, indicating that KLF2 knockdown suppressed the megakaryocytic differentiation. Further, combination of the RNA-seq and ChIP-seq results suggested that chimerin 1 (CHN1) and potassium voltage-gated channel subfamily Q member 5 (KCNQ5) may be target genes regulated of KLF2. Both CHN1 and KCNQ5 knockdown could block the megakaryocytic differentiation to some content. CONCLUSION: This study implicated a regulatory role of KLF2 in megakaryocytic differentiation, which may suggest KLF2 as a target for illness with abnormal megakaryocytic differentiation.
The involvement of krüppel-like transcription factor 2 in megakaryocytic differentiation induction by phorbol 12-myrestrat 13-acetate.
佛波醇 12-肉豆蔻酸酯 13-乙酸酯诱导巨核细胞分化过程中 Krüppel 样转录因子 2 的参与
阅读:27
| 期刊: | Biomarker Research | 影响因子: | 11.500 |
| 时间: | 2024 | 起止号: | 2024 Jul 17; 12(1):65 |
| doi: | 10.1186/s40364-024-00614-9 | 种属: | Rat |
| 研究方向: | 细胞生物学 | ||
文献解析
1. 领域背景与文献引入
文献英文标题:The involvement of krüppel-like transcription factor 2 in megakaryocytic differentiation induction by phorbol 12-myrestrat 13-acetate;发表期刊:Biomarker Research;影响因子:未公开;研究领域:巨核细胞分化调控。
巨核细胞分化是造血干细胞向血小板生成的关键步骤,其过程由造血干细胞经巨核细胞-红细胞祖细胞(MEP)、集落形成单位-巨核细胞(CFU-MK)逐步成熟为巨核细胞,最终释放血小板。该过程的异常会导致血小板生成障碍,如异基因造血干细胞移植(allo-HSCT)后常见的持续性血小板减少症(PT),其发病率达5%-37%,严重影响患者预后。当前研究热点聚焦于转录因子在巨核细胞分化中的时空调控作用,其中克罗姆样转录因子家族(KLFs)因参与造血细胞增殖与分化而成为研究重点。已有研究表明,KLF1调控红细胞成熟、KLF4调控造血干细胞功能,但KLF2在巨核细胞分化中的功能及机制尚未明确。针对这一研究空白,本研究旨在探究KLF2在佛波酯12-肉豆蔻酸酯13-乙酸酯(PMA,巨核细胞分化诱导剂)诱导的巨核细胞分化中的作用,并解析其分子机制,为血小板减少症的靶向干预提供新的理论依据。
2. 文献综述解析
文献综述围绕“巨核细胞分化的转录调控”与“KLF2的功能研究现状”展开逻辑评述。作者首先总结巨核细胞分化的复杂性:受多种转录因子时空调控,如GATA-1、NF-E2等;随后介绍KLF家族的共性特征(C2H2型锌指结构、结合GC-rich DNA序列)及功能多样性,如KLF1调控红细胞成熟、KLF4调控干细胞自我更新;进一步指出KLF2在其他细胞分化中的作用,如促进胚胎红细胞前体细胞成熟、内皮细胞分化,但KLF2在巨核细胞分化中的角色尚未报道。现有研究的关键结论是KLF家族是造血分化的重要调控因子,但KLF2在巨核细胞中的功能缺失;技术方法的优势在于利用公共数据库(BloodSpot、GEO)筛选差异基因,结合细胞模型验证,但局限性是缺乏KLF2在巨核细胞中的功能及机制研究。本研究的创新点在于首次明确KLF2在PMA诱导的巨核细胞分化中的促进作用,并通过组学技术鉴定其直接靶基因CHN1和KCNQ5,填补了KLF2在巨核细胞分化调控中的研究空白。
3. 研究思路总结与详细解析
本研究的整体框架为“数据库筛选→细胞模型验证→功能实验→组学解析→靶基因验证”,旨在回答“KLF2是否调控巨核细胞分化?如何调控?”的核心科学问题。技术路线以PMA诱导的K562/HEL细胞巨核细胞分化模型为基础,通过慢病毒调控KLF2表达,结合流式细胞术、形态学染色、蛋白质免疫印迹(Western blot)验证功能,再利用RNA测序(RNA-seq)和染色质免疫沉淀测序(ChIP-seq)鉴定靶基因,最终通过小干扰RNA(siRNA)验证靶基因功能。
3.1 KLF家族在巨核细胞分化中的表达模式分析
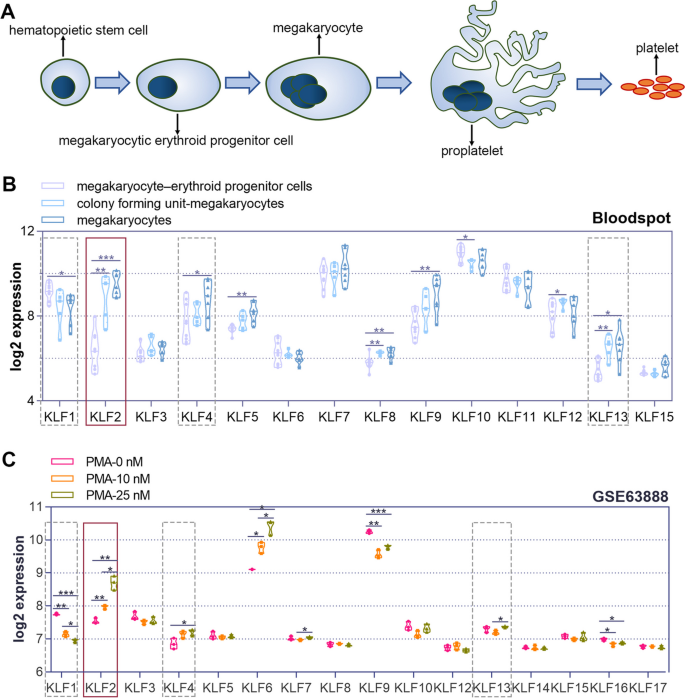
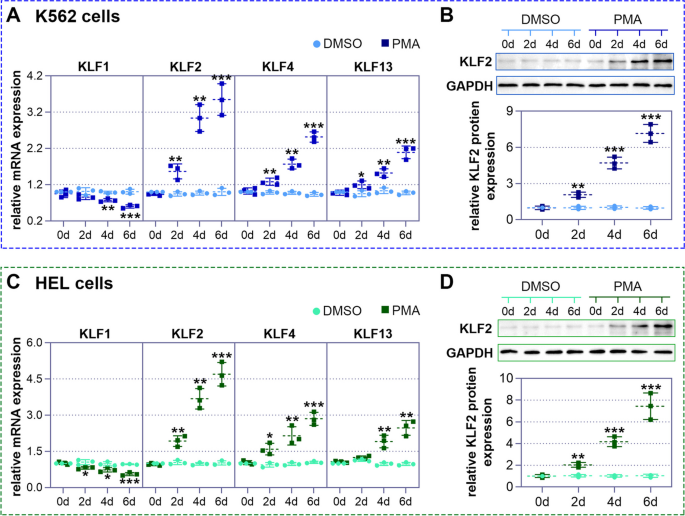
实验目的是筛选巨核细胞分化过程中差异表达的KLF家族成员。方法上,首先从BloodSpot数据库获取正常造血细胞(MEP、CFU-MK、巨核细胞)的KLFs表达数据,从GEO数据库(GSE63888)获取PMA处理K562细胞的KLFs表达数据;随后用25 nM PMA处理K562和HEL细胞(0、2、4、6天),通过实时聚合酶链式反应(实时PCR)检测KLF1、KLF2、KLF4、KLF13的mRNA水平,Western blot检测KLF2的蛋白水平。结果显示,KLF2在正常巨核细胞中的表达高于MEP和CFU-MK(BloodSpot数据库);PMA处理K562细胞6天后,KLF2 mRNA水平较0天升高约3倍,蛋白水平升高约2.5倍(n=3,P<0.01);HEL细胞中呈现类似趋势(图1、图2)。实验所用关键产品:KLF2抗体(Thermofisher,货号PA5-40591)、实时PCR引物(定制)。
3.2 KLF2对巨核细胞分化的功能验证
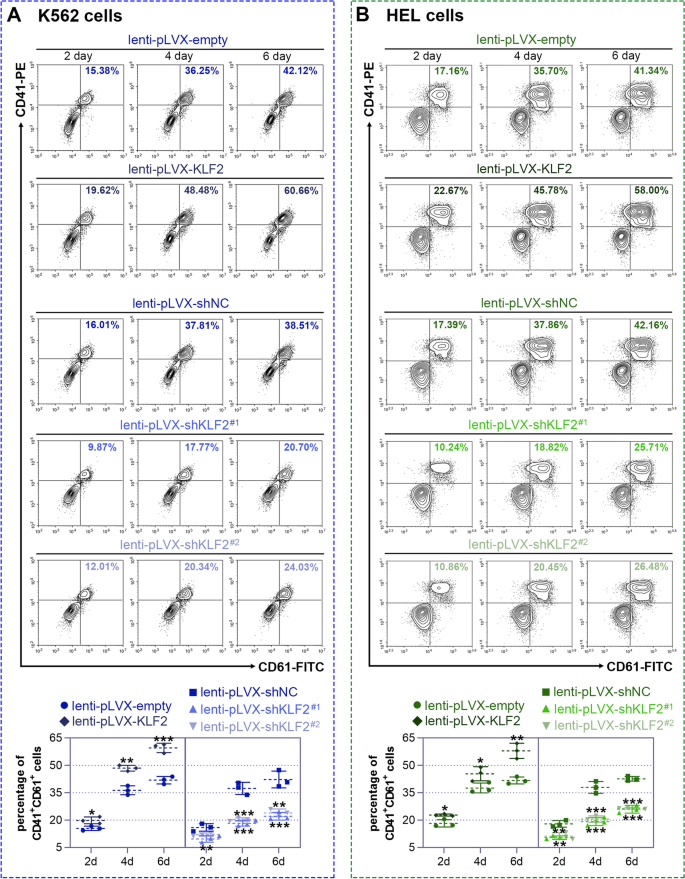
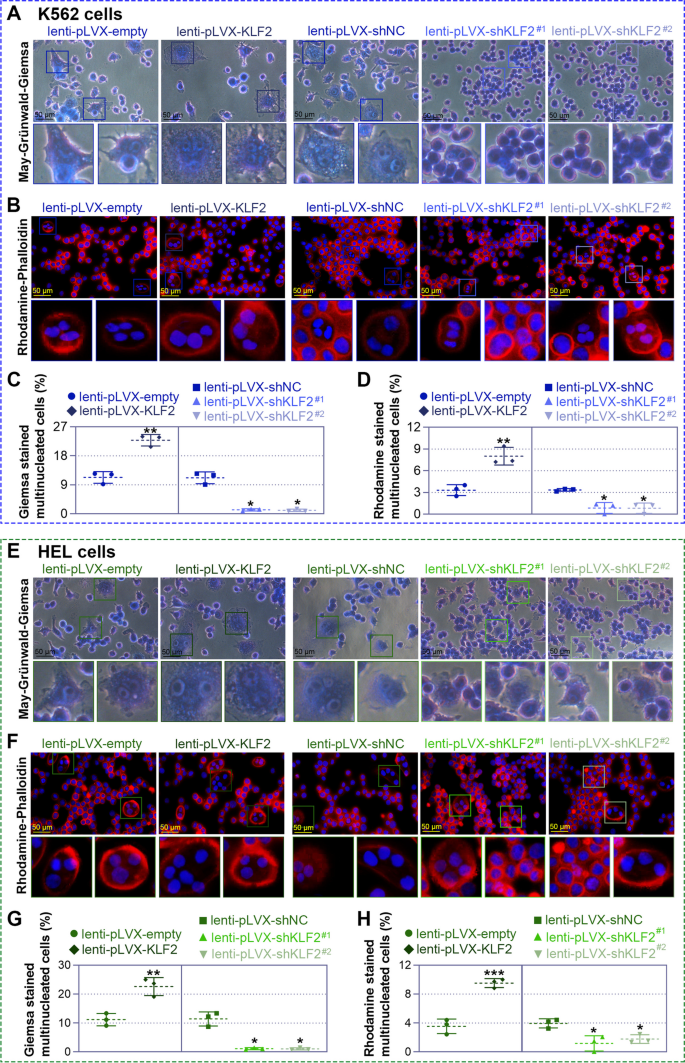
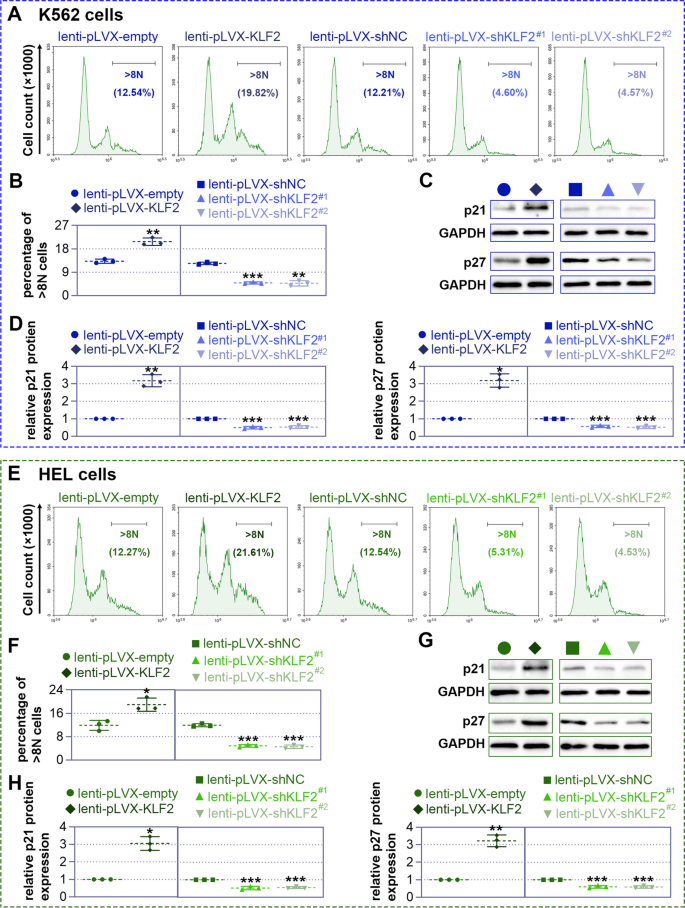
实验目的是明确KLF2对巨核细胞分化的调控作用。方法上,通过慢病毒载体构建KLF2过表达(lenti-pLVX-KLF2)和敲低(lenti-pLVX-shKLF2#1/#2)细胞模型,感染K562/HEL细胞72小时后,用25 nM PMA诱导分化;通过流式细胞术检测巨核细胞表面标志物CD41(血小板膜糖蛋白Ⅱb)/CD61(血小板膜糖蛋白Ⅲa)的阳性细胞比例,May-Grünwald-Giemsa染色观察细胞形态,鬼笔环肽染色检测多核化,碘化丙啶(PI)染色流式检测多倍体(>8N)比例,Western blot检测分化相关蛋白p21、p27的表达。结果显示,过表达KLF2使K562细胞CD41/CD61阳性比例较对照组增加约1.8倍(6天,n=3,P<0.05),多核细胞数增加约2倍,>8N细胞比例升高约1.5倍,p21、p27蛋白水平升高约2倍;敲低KLF2则呈现相反结果(图3、图4、图5)。实验所用关键产品:CD41抗体(Thermo Fisher)、CD61抗体(Proteintech)、p21抗体(ABclonal,货号A19094)、p27抗体(ABclonal,货号A5357)。
3.3 KLF2靶基因的鉴定与验证
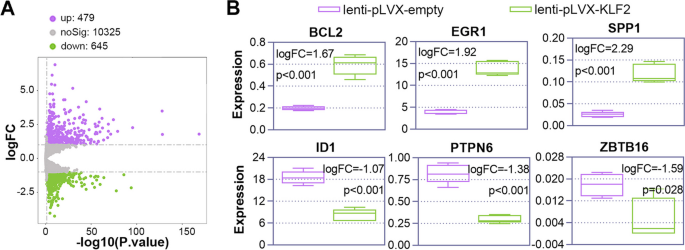
实验目的是解析KLF2调控巨核细胞分化的分子机制。方法上,对KLF2过表达的K562细胞进行RNA-seq(筛选差异表达基因:p<0.05、|log2折叠变化|>1)和ChIP-seq(检测KLF2结合的启动子区域),通过Venn图交集筛选KLF2的直接靶基因;随后用siRNA敲低靶基因Chimerin 1(CHN1)和钾电压门控通道Q亚家族成员5(KCNQ5),检测CD41/CD61阳性细胞比例。结果显示,RNA-seq筛选到1124个差异基因(479上调、645下调),ChIP-seq鉴定到75个KLF2结合的启动子区域基因,两者交集得到CHN1和KCNQ5(图6、图7);敲低CHN1或KCNQ5使K562细胞CD41/CD61阳性比例较对照组降低约40%(n=3,P<0.05)(图8)。实验所用关键产品:RNA-seq试剂盒(Illumina HiSeqTM)、ChIP-seq试剂盒(Illumina NovaSeq 6000)、CHN1 siRNA(JTS Scientific)、KCNQ5 siRNA(JTS Scientific)。


4. Biomarker研究及发现成果解析
本研究涉及的生物标志物(Biomarker)包括“调控型Biomarker”KLF2及“靶标型Biomarker”CHN1、KCNQ5。KLF2作为转录因子,是巨核细胞分化的关键调控节点;CHN1和KCNQ5作为KLF2的直接靶基因,介导其促分化作用。
Biomarker定位与筛选逻辑
KLF2的筛选逻辑为“公共数据库(BloodSpot、GEO)差异分析→细胞模型(K562/HEL)验证→功能实验确认”;CHN1和KCNQ5的筛选逻辑为“RNA-seq差异基因→ChIP-seq结合启动子区域→Venn图交集→siRNA功能验证”,形成完整的“筛选-验证”链条。
研究过程与数据
KLF2的来源为正常造血细胞(BloodSpot数据库)和PMA诱导的细胞模型(K562/HEL),验证方法包括实时PCR(mRNA水平)、Western blot(蛋白水平)及慢病毒调控后的功能实验;CHN1和KCNQ5的来源为组学联合分析的交集基因,验证方法为siRNA敲低后的流式检测。数据显示:1)BloodSpot数据库中,巨核细胞KLF2表达较MEP高约1.6倍,较CFU-MK高约1.3倍;2)PMA处理K562细胞6天后,KLF2蛋白水平较0天升高约2.5倍(n=3,P<0.01);3)过表达KLF2使CD41/CD61阳性比例较对照组增加约1.8倍(n=3,P<0.05);4)敲低CHN1或KCNQ5使CD41/CD61阳性比例降低约40%(n=3,P<0.05)。
核心成果
1)功能关联:KLF2是巨核细胞分化的正调控因子,过表达促进分化、敲低抑制分化;2)分子机制:CHN1和KCNQ5是KLF2的直接靶基因,介导其促分化作用;3)临床意义:KLF2可能作为血小板减少症的潜在干预靶点,通过上调KLF2或其靶基因促进巨核细胞分化,恢复血小板生成。
本研究的创新性在于首次将KLF2与巨核细胞分化关联,并鉴定其靶基因,为巨核细胞分化的分子调控网络提供了新的节点,也为血小板减少症的靶向治疗提供了新的理论依据。
特别声明
1、本页面内容包含部分的内容是基于公开信息的合理引用;引用内容仅为补充信息,不代表本站立场。
2、若认为本页面引用内容涉及侵权,请及时与本站联系,我们将第一时间处理。
3、其他媒体/个人如需使用本页面原创内容,需注明“来源:[生知库]”并获得授权;使用引用内容的,需自行联系原作者获得许可。
4、投稿及合作请联系:info@biocloudy.com。