1. 领域背景与文献引入
文献英文标题:Endoplasmic reticulum stress response in an INS-1 pancreatic β-cell line with inducible expression of a folding-deficient proinsulin;发表期刊:BMC Cell Biology;影响因子:未公开;研究领域:胰腺β细胞内质网应激与糖尿病
领域共识:2型糖尿病的核心病理特征是胰腺β细胞功能障碍与凋亡,导致胰岛素分泌不足。生理状态下,餐后高血糖刺激胰岛素合成与分泌,会短暂增加β细胞内质网的蛋白折叠负荷,诱导一过性内质网应激;病理状态下,肥胖、持续高糖、游离脂肪酸积累等因素会引发慢性内质网应激,激活未折叠蛋白反应(UPR),若应激持续存在则会触发β细胞凋亡,这是2型糖尿病进展的关键机制之一。领域发展关键节点包括1999年Akita小鼠模型的发现,该模型携带胰岛素2基因C96Y突变,导致前胰岛素折叠缺陷并滞留内质网,引发内质网应激与β细胞凋亡,为研究遗传因素导致的β细胞内质网应激提供了体内模型。当前研究热点聚焦于解析β细胞内质网应激的特异性调控机制,以及寻找保护β细胞免受内质网应激损伤的靶点,但未解决的核心问题包括:缺乏可精准调控的β细胞内质网应激模型,无法模拟生理病理下突变蛋白积累的时间动态过程;β细胞作为高分泌细胞,其内质网应激的基因表达谱与非β细胞的差异尚不明确;内质网相关降解(ERAD)系统在β细胞中的具体保护机制仍需深入解析。
针对上述研究空白,本研究基于Akita小鼠的突变机制,构建了可诱导表达折叠缺陷型胰岛素2-EGFP融合蛋白的INS-1β细胞系,实现了β细胞内质网应激的时间可控诱导,系统解析了不同时间点的基因表达响应规律,并验证了ERAD系统在维持β细胞存活中的关键作用,为β细胞内质网应激的机制研究提供了特异性模型与新的靶点。
2. 文献综述解析
作者对领域内现有研究的分类维度为内质网应激的诱导方式(生理刺激、病理毒素、遗传突变)与细胞类型(β细胞、非β细胞)。现有研究的关键结论包括:内质网应激通过PERK、IRE1、ATF6三条保守通路激活未折叠蛋白反应,在多数细胞中,未折叠蛋白反应先通过抑制全局翻译、上调内质网伴侣蛋白来恢复内质网稳态,若应激持续则触发凋亡;技术方法优势方面,毒素诱导模型(如毒胡萝卜素、衣霉素)能快速、高效激活内质网应激,适用于急性应激机制研究;Akita小鼠模型能模拟遗传突变导致的慢性内质网应激,适用于体内病理机制研究;但现有研究存在明显局限性:毒素诱导模型缺乏生理相关性,无法模拟突变蛋白积累的病理过程;体内模型难以进行时间动态的分子机制解析;且多数研究聚焦于非β细胞的内质网应激响应,β细胞作为高分泌细胞,其未折叠蛋白反应的特异性基因谱尚未明确,ERAD系统在β细胞中的细胞保护机制也未被系统验证。
通过对比现有研究的未解决问题,本研究的创新价值凸显:首次构建了基于遗传突变的可诱导β细胞内质网应激模型,实现了应激过程的时间可控性,弥补了毒素诱导模型与体内模型的不足;系统解析了β细胞内质网应激的时间动态基因表达谱,鉴定了β细胞特异性的内质网应激响应基因,完善了领域内对β细胞未折叠蛋白反应的认知;明确了ERAD系统尤其是Herp蛋白在维持β细胞存活中的关键作用,为2型糖尿病中β细胞保护提供了新的潜在靶点。
3. 研究思路总结与详细解析
本研究的整体框架为:以构建可诱导表达折叠缺陷型胰岛素的β细胞模型为基础,验证内质网应激通路的激活状态,通过时间动态转录组分析解析基因表达响应规律,检测内质网应激诱导的凋亡,最终验证ERAD系统的细胞保护功能,形成“模型构建→应激验证→转录分析→凋亡检测→机制验证”的完整研究闭环。研究目标是解析β细胞内质网应激的时间动态响应及ERAD的细胞保护机制,核心科学问题是折叠缺陷型胰岛素如何激活β细胞的未折叠蛋白反应通路,以及ERAD系统如何调控β细胞存活。
3.1 可诱导折叠缺陷型胰岛素表达INS-1细胞系构建与表征
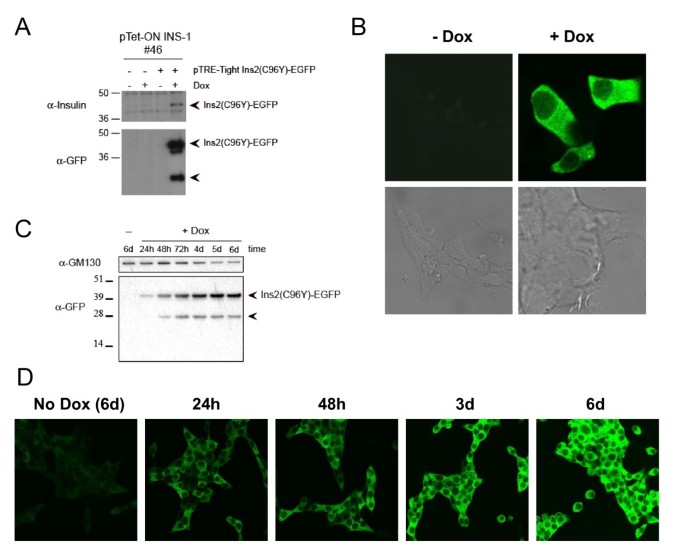
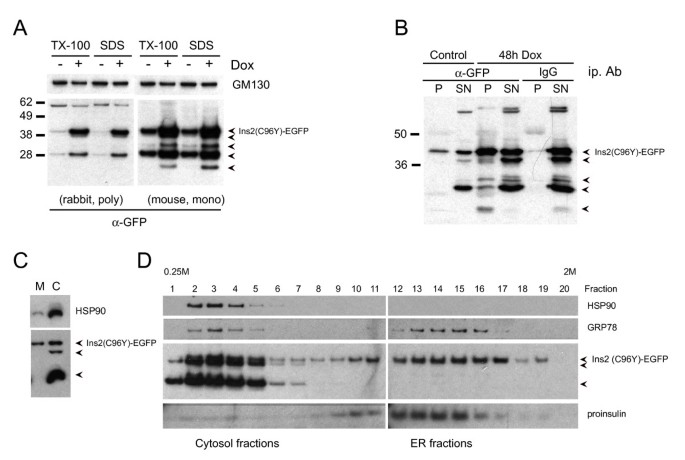
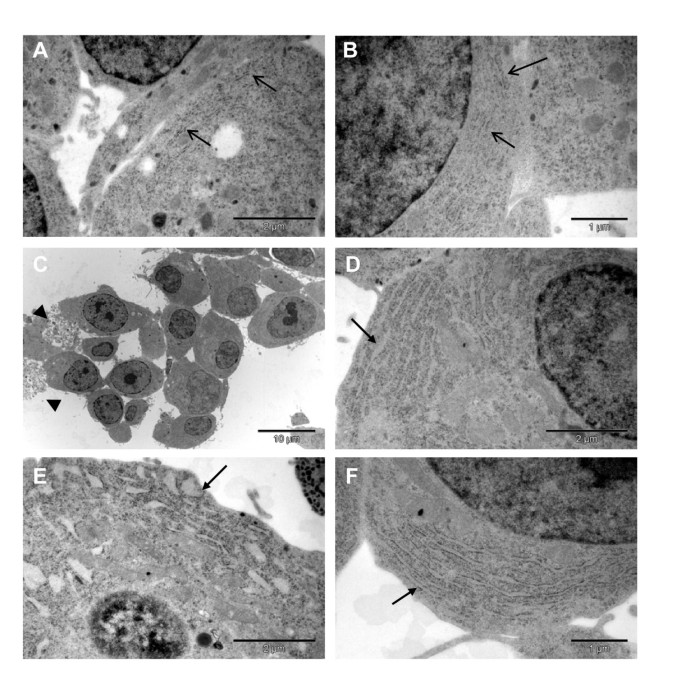
实验目的为构建基于Akita小鼠C96Y突变的可诱导表达胰岛素2-EGFP融合蛋白的INS-1细胞系,模拟β细胞内质网应激的病理过程。方法细节包括:首先通过转染pTet-ON质粒并筛选,获得可响应强力霉素诱导表达的INS-1稳定细胞系;随后通过定点突变构建携带C96Y突变的胰岛素2 cDNA,与EGFP融合后插入pTRE-Tight载体,将该载体与潮霉素抗性质粒共转染入pTet-ON INS-1细胞,经G418与潮霉素双重筛选获得双稳定细胞系;通过流式细胞术分选强力霉素诱导48h后EGFP高表达的细胞(前20%),得到#4S2克隆,后续实验采用2μg/ml强力霉素诱导融合蛋白表达。结果解读显示,Western blot与荧光显微镜检测表明,强力霉素处理后Ins2(C96Y)-EGFP融合蛋白表达显著上调,且定位于细胞质(图1);进一步的细胞组分分离实验显示,全长融合蛋白定位于内质网,降解产物仅存在于胞质(图2);电子显微镜观察发现,未诱导细胞的内质网形态正常,诱导3天后内质网出现扩张,6天后扩张更为显著,且部分细胞呈现凋亡形态(图3),表明融合蛋白的成功表达诱导了β细胞内质网应激。实验所用关键产品:Invitrogen的QuikChange II XL定点突变试剂盒、pCRII-TOPO载体、潮霉素B、G418、TRIzol试剂;Clontech的pTet-ON质粒、pTRE-Tight载体;BD Biosciences的pEGFP-N1载体;Cell Signaling的Phospho-eIF2α抗体;Santa Cruz的胰岛素抗体;Sigma的γ-tubulin抗体等。
3.2 内质网应激通路激活验证
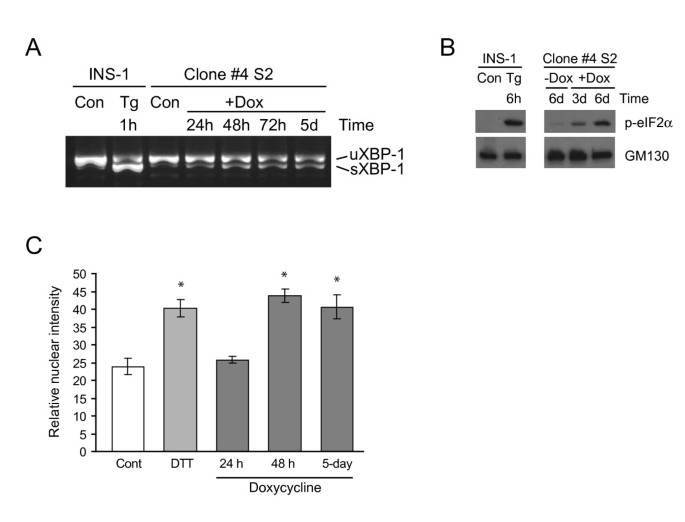
实验目的为验证Ins2(C96Y)-EGFP表达是否激活β细胞的三条经典未折叠蛋白反应通路(PERK、IRE1、ATF6)。方法细节包括:用2μg/ml强力霉素诱导#4S2细胞不同时间(24h、48h、5天等),通过RT-PCR检测XBP-1的剪接情况(IRE1通路激活的标志),Western blot检测磷酸化eIF2α的水平(PERK通路激活的标志),免疫荧光染色结合ImageJ定量分析核内ATF6的荧光强度(ATF6通路激活的标志)。结果解读显示,RT-PCR结果表明,诱导24h后即可检测到剪接型XBP-1,且该状态持续至诱导7天(图4A);Western blot结果显示,磷酸化eIF2α的水平随诱导时间增加而显著升高(图4B);免疫荧光定量结果显示,诱导后核内ATF6的荧光强度显著高于未诱导组(图4C),表明三条未折叠蛋白反应通路均被成功激活,证实了模型的有效性。实验所用关键产品:Applied Biosystems的TaqMan基因表达引物;Cell Signaling的Phospho-eIF2α抗体;定制的anti-ATF6抗体;Zeiss激光扫描共聚焦显微镜等。
3.3 时间动态基因表达谱分析
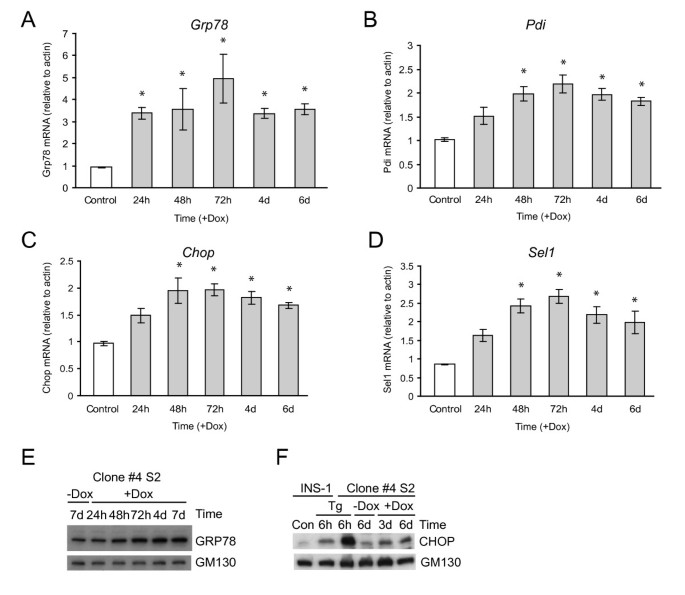
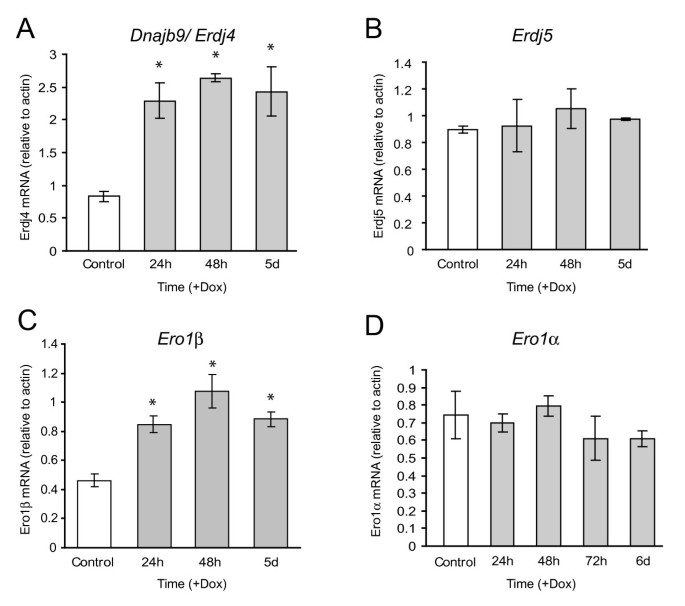
实验目的为解析Ins2(C96Y)-EGFP诱导后不同时间点的基因表达变化,明确β细胞内质网应激的转录响应规律。方法细节包括:分别在诱导24h、48h、5天后提取细胞总RNA,进行Affymetrix Rat Genome 230 2.0芯片分析,筛选差异表达基因(筛选标准为:与未诱导组相比表达变化≥2倍,且在至少2次重复实验中显著);随后采用实时荧光定量PCR对芯片中未检测到的关键内质网应激基因(如Grp78、Pdi)进行验证。结果解读显示,芯片分析结果表明,诱导24h时有45个基因上调、5个基因下调;诱导48h时有86个基因上调、37个基因下调;诱导5天时68个基因上调、56个基因下调。时间动态分析显示,诱导24h时主要上调的基因包括内质网伴侣蛋白基因(Erdj4、P58IPK、Erdj3、Fkbp11)与内质网相关降解基因(Herp、Sel1);诱导48h时,凋亡相关转录因子基因Chop与Trib3的上调幅度显著增加;实时荧光定量PCR验证了Grp78、Pdi等芯片未检测到的上调基因,证实了芯片分析的可靠性(图5、6)。实验所用关键产品:Affymetrix Rat Genome 230 2.0芯片;Applied Biosystems的High-Capacity cDNA Reverse Transcription Kit、TaqMan基因表达系统;Qiagen的RNeasy Mini Kit等。
3.4 内质网应激诱导的凋亡检测
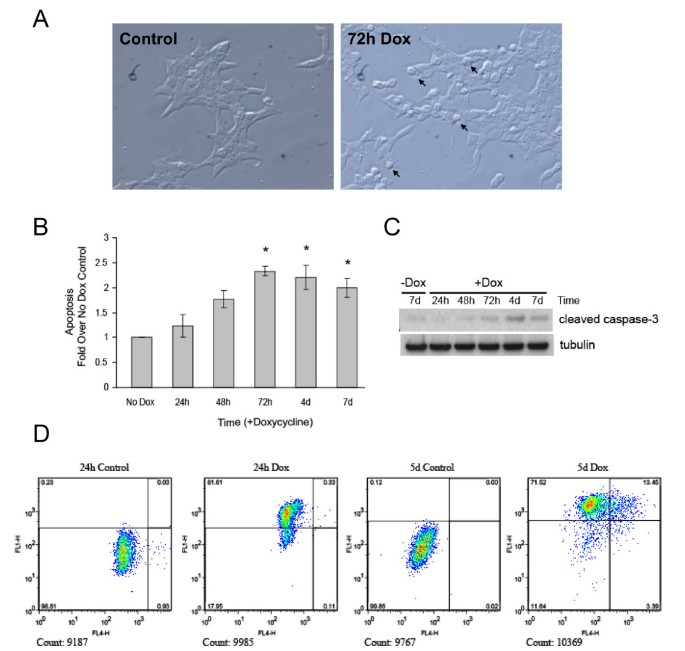
实验目的为检测Ins2(C96Y)-EGFP表达是否诱导β细胞凋亡,并分析凋亡的时间依赖性。方法细节包括:通过光镜观察细胞形态变化;采用细胞死亡检测ELISA试剂盒检测胞质中DNA-组蛋白复合物的水平(凋亡的标志);Western blot检测cleaved caspase-3的表达;通过TUNEL染色结合流式细胞术,同时检测EGFP表达(融合蛋白表达标志)与TUNEL阳性(凋亡标志)的细胞比例。结果解读显示,光镜观察发现,诱导72h后部分细胞形态变圆,呈现凋亡特征(图7A);细胞死亡ELISA结果表明,凋亡水平随诱导时间增加而显著升高(n=4,P<0.05,图7B);Western blot结果显示,诱导3天后cleaved caspase-3的表达水平显著上调(图7C);流式细胞术结果显示,诱导5天时约14%的EGFP阳性细胞为TUNEL阳性,表明融合蛋白表达诱导了β细胞凋亡(图7D)。实验所用关键产品:Roche的细胞死亡检测ELISA试剂盒、APO-BrdU TUNEL Assay Kit;BD的FACSCalibur流式细胞仪;Cell Signaling的cleaved caspase-3抗体等。
3.5 ERAD系统的细胞保护功能验证
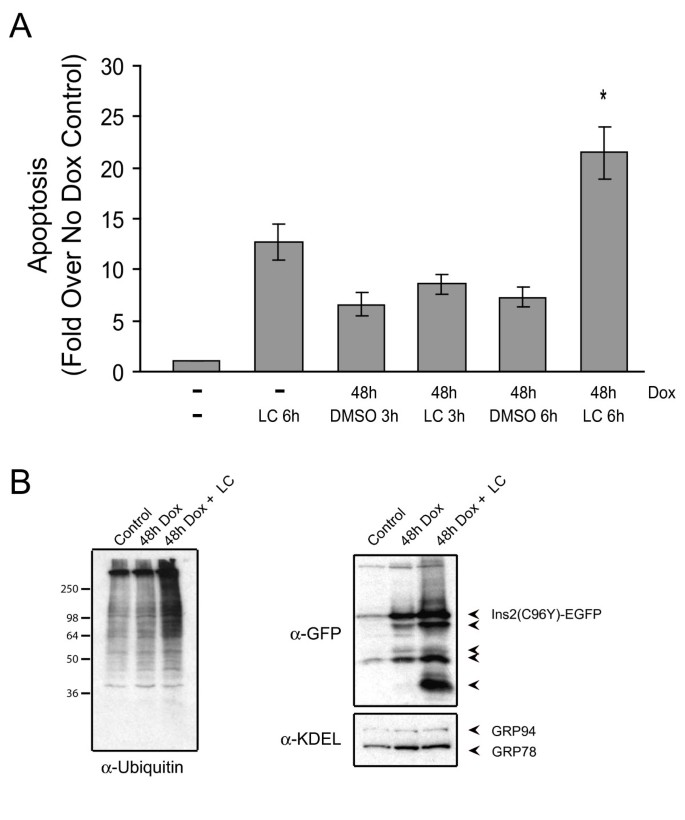
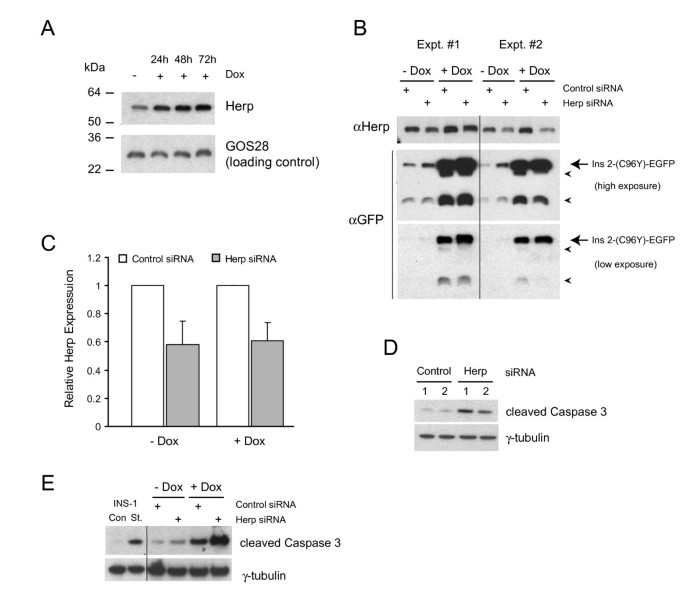
实验目的为验证内质网相关降解系统在维持β细胞存活中的作用,明确Herp蛋白的具体功能。方法细节包括:采用蛋白酶体抑制剂乳胞素处理诱导后的细胞,抑制内质网相关降解功能,检测凋亡水平与Ins2(C96Y)-EGFP融合蛋白的积累;采用siRNA敲低Herp蛋白的表达,检测融合蛋白的水平变化与细胞凋亡情况。结果解读显示,乳胞素处理后,凋亡细胞比例显著高于对照组(n=3,P<0.05,图8A),同时细胞内泛素化蛋白与Ins2(C96Y)-EGFP融合蛋白的水平显著升高(图8B);Herp siRNA敲低实验显示,Herp蛋白的表达被敲低40-50%(n=4),同时Ins2(C96Y)-EGFP融合蛋白的水平显著升高,细胞凋亡水平也显著增加(图9),表明内质网相关降解系统通过降解折叠缺陷型胰岛素,维持β细胞的存活,而Herp蛋白在该过程中发挥关键作用。实验所用关键产品:Invitrogen的Herp siRNA、Lipofectamine RNAiMAX;Roche的完全蛋白酶抑制剂混合物;定制的Herp抗体等。
4. Biomarker研究及发现成果解析
本研究未聚焦传统的疾病诊断型生物标志物,而是鉴定了β细胞内质网应激响应的特异性基因生物标志物,其筛选逻辑为:通过时间动态转录组分析,对比非β细胞的内质网应激基因表达谱,筛选仅在β细胞中被折叠缺陷型胰岛素诱导上调的基因,并通过功能实验验证其与β细胞存活的关联。
研究过程详述包括:通过Affymetrix芯片与实时荧光定量PCR,鉴定了Erdj4、P58IPK、Erdj3、Fkbp11等β细胞特异性上调的内质网伴侣蛋白基因,以及Sdf2l1、Armet、Creld2等在诱导后持续高表达的基因;验证方法包括实时荧光定量PCR(检测mRNA水平)与Western blot(检测蛋白水平);特异性分析显示,这些基因在β细胞中被折叠缺陷型胰岛素诱导上调,但在神经母细胞瘤、星形胶质细胞等非β细胞的毒素诱导内质网应激中未被诱导,且在棕榈酸诱导的MIN6β细胞内质网应激中显著上调,与糖尿病模型胰岛中的表达趋势一致;敏感性方面,这些基因在诱导24h后即显著上调,可作为β细胞内质网应激的早期响应标志物。
核心成果提炼包括:首次鉴定了β细胞内质网应激响应的特异性基因谱,为β细胞内质网应激的检测提供了潜在的分子标志物;明确了内质网相关降解基因Herp在维持β细胞存活中的关键作用,敲低Herp表达后β细胞凋亡水平显著升高(n=4,P<0.05),提示Herp可作为保护β细胞免受内质网应激损伤的潜在靶点;本研究构建的可诱导细胞模型为后续筛选β细胞内质网应激的特异性生物标志物与治疗靶点提供了重要工具。