Background
Huntington's disease (HD) is a neurodegenerative disease that involves a complex combination of psychiatric, cognitive and motor impairments. Synaptic dysfunction has been implicated in HD pathogenesis. However, the mechanisms have not been clearly delineated. Synaptic vesicular zinc is closely linked to modulating synaptic transmission and maintaining cognitive ability. It is significant to assess zinc homeostasis for further revealing the pathogenesis of synaptic dysfunction and cognitive impairment in HD.
Conclusions
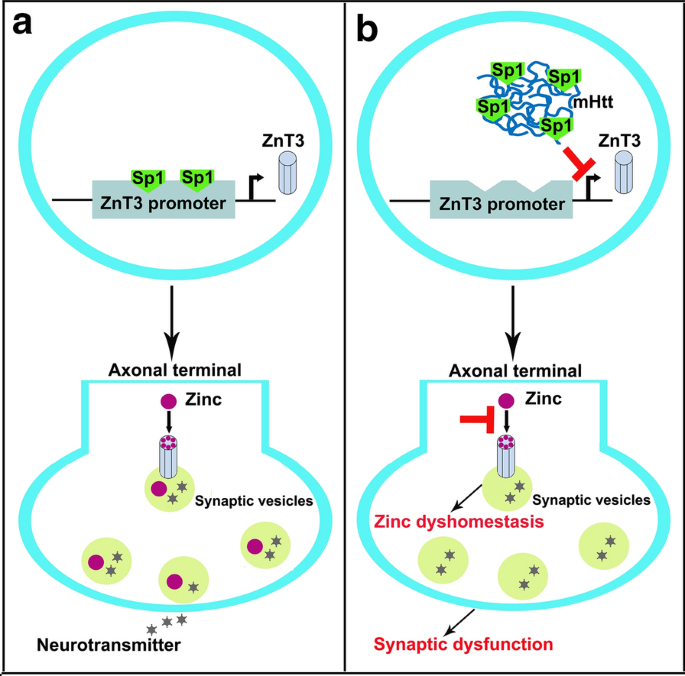
This is first study to reveal a significant loss of synaptic vesicular zinc and a decline in ZnT3 transcriptional activity in the HD transgenic mice. Our work sheds a novel mechanistic insight into pathogenesis of HD that mutant huntingtin down-regulates expression of ZnT3 through inhibiting binding of Sp1 to the promoter of ZnT3 gene, causing disruption of synaptic vesicular zinc homeostasis. Disrupted vesicular zinc ultimately leads to early synaptic dysfunction and cognitive deficits in HD. It is also suggested that maintaining normal synaptic vesicular zinc concentration is a potential therapeutic strategy for HD.
Results
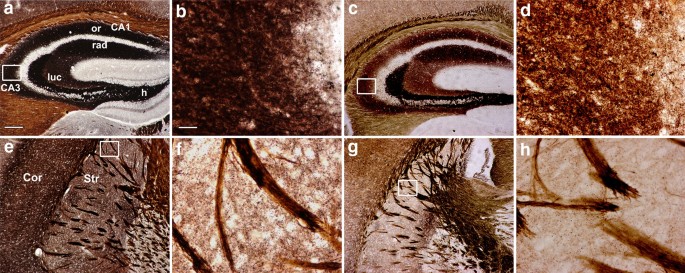
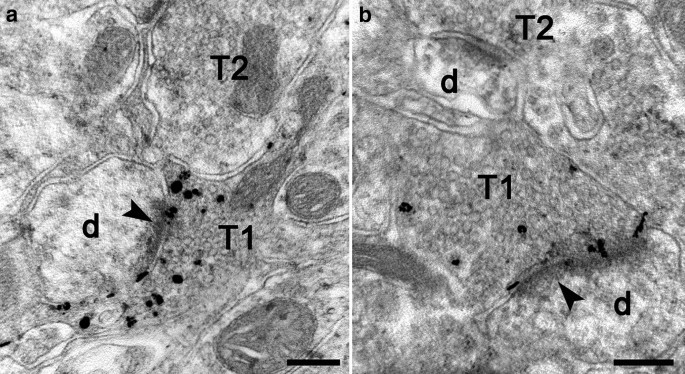
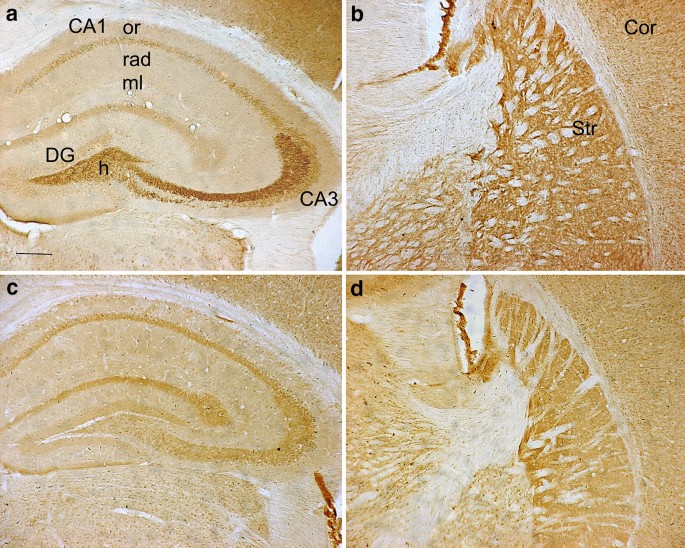
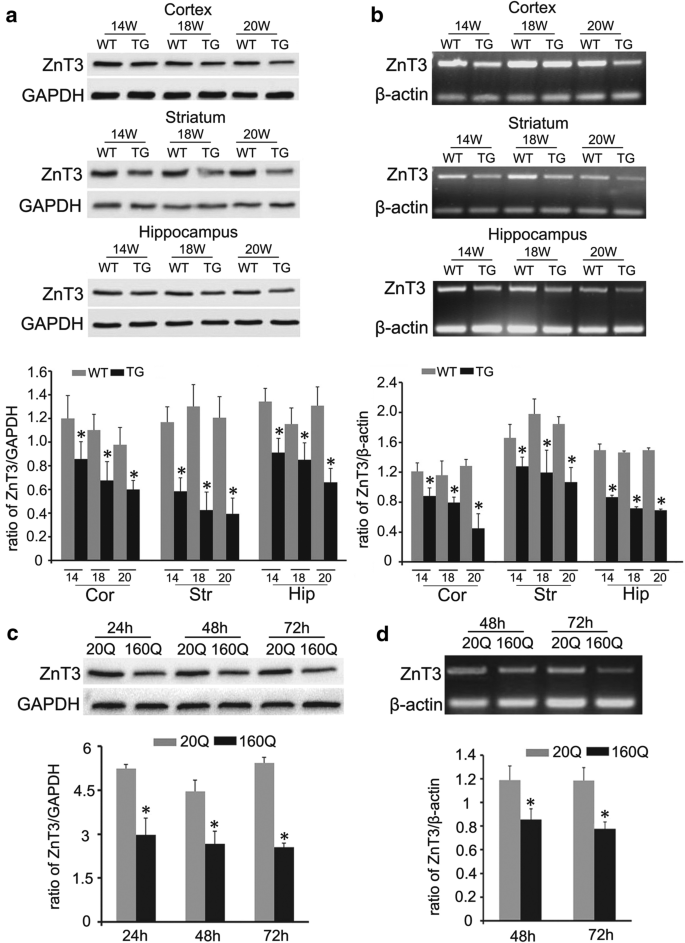
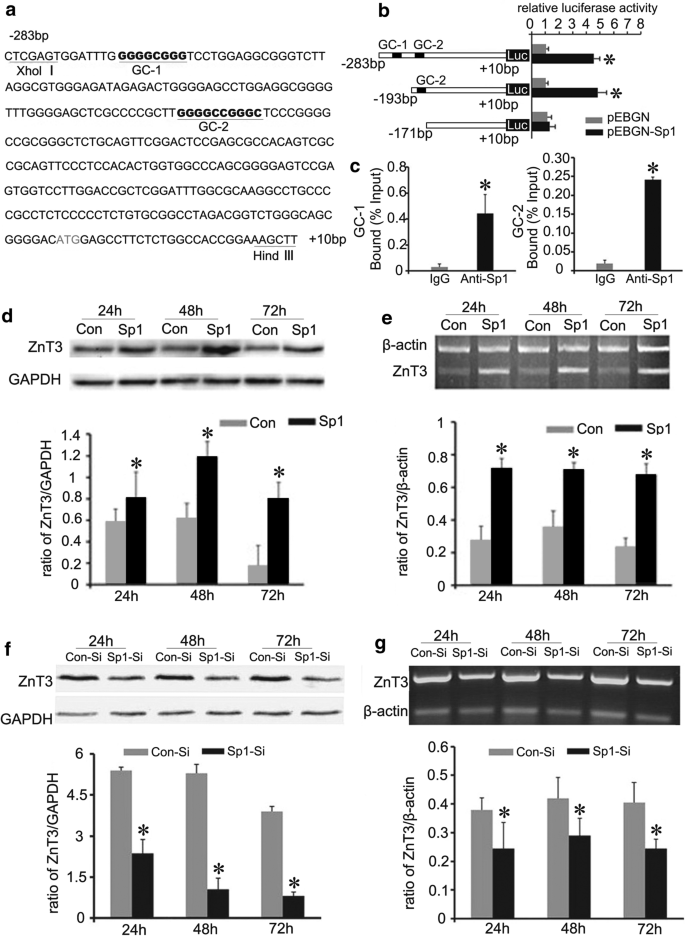
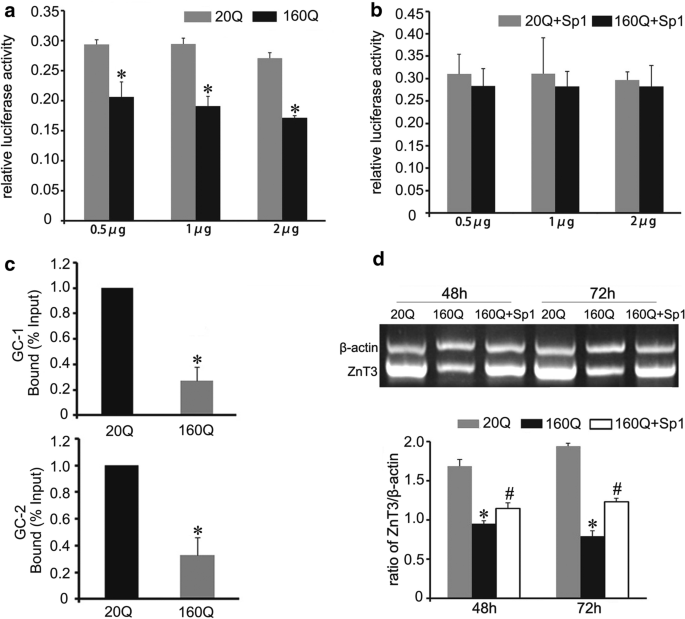
Histochemical staining by autometallography indicated that synaptic vesicular zinc was decreased in the hippocampus, cortex and striatum of N171-82Q HD transgenic mice. Analyses by immunohistochemistry, Western blot and RT-PCR found that the expression of zinc transporter 3 (ZnT3) required for transport of zinc into synaptic vesicles was obviously reduced in these three brain regions of the HD mice aged from 14 to 20 weeks and BHK cells expressing mutant huntingtin. Significantly, dual-luciferase reporter gene and chromatin immunoprecipitation assays demonstrated that transcription factor Sp1 could activate ZnT3 transcription via its binding to the GC boxes in ZnT3 promoter. Moreover, mutant huntingtin was found to inhibit the binding of Sp1 to the promoter of ZnT3 and down-regulate ZnT3 expression, and the decline in ZnT3 expression could be ameliorated through overexpression of Sp1. Conclusions: This is first study to reveal a significant loss of synaptic vesicular zinc and a decline in ZnT3 transcriptional activity in the HD transgenic mice. Our work sheds a novel mechanistic insight into pathogenesis of HD that mutant huntingtin down-regulates expression of ZnT3 through inhibiting binding of Sp1 to the promoter of ZnT3 gene, causing disruption of synaptic vesicular zinc homeostasis. Disrupted vesicular zinc ultimately leads to early synaptic dysfunction and cognitive deficits in HD. It is also suggested that maintaining normal synaptic vesicular zinc concentration is a potential therapeutic strategy for HD.