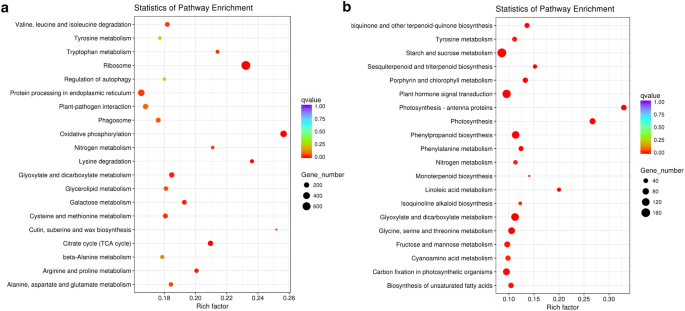
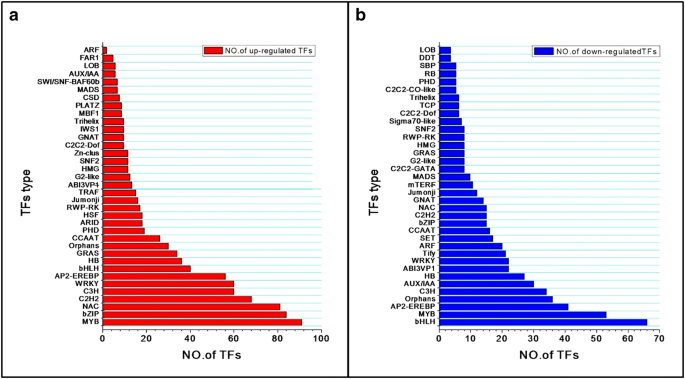
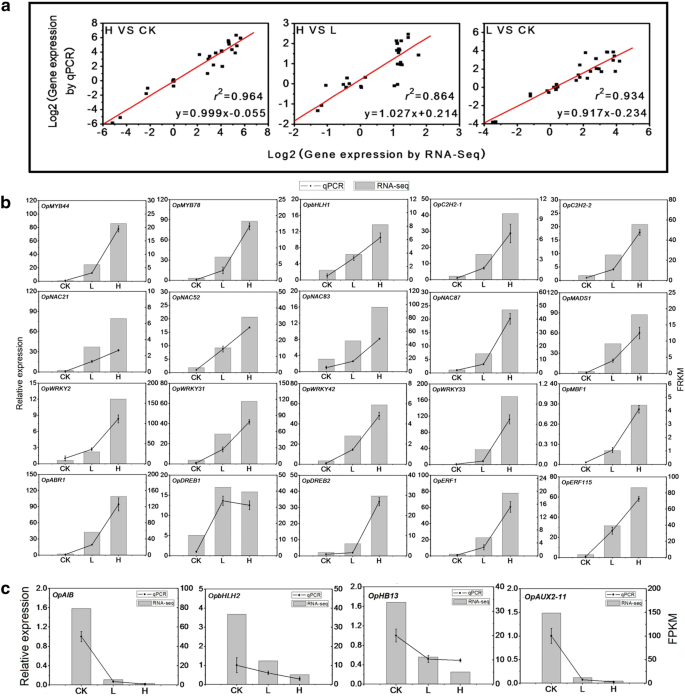
Abstract
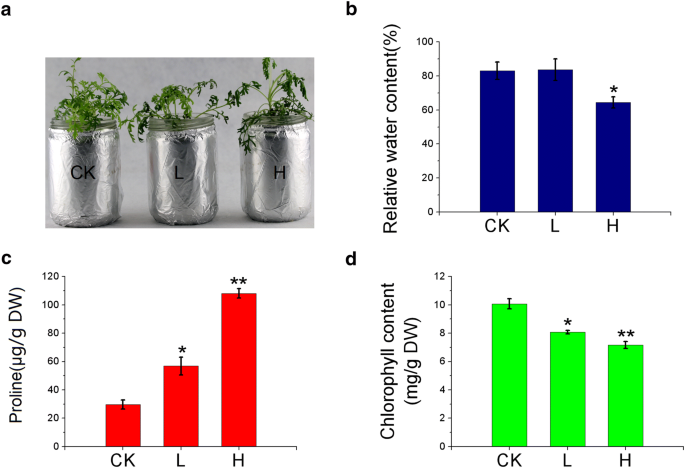
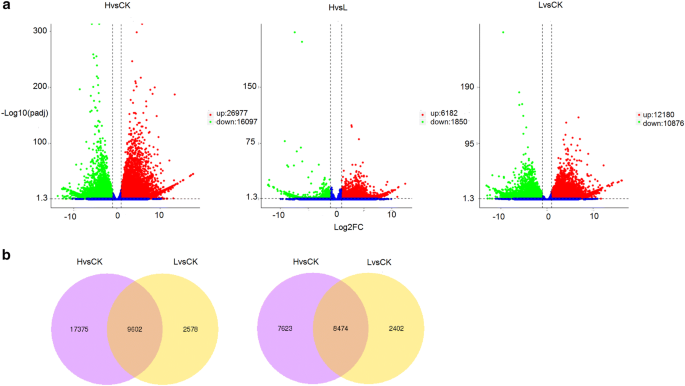
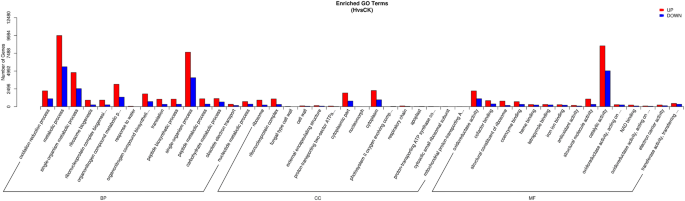
BACKGROUND: Water scarcity is considered to be a severe environmental constraint to plant survival and productivity. Studies on drought-tolerant plants would definitely promote a better understanding of the regulatory mechanism lying behind the adaptive response of plants to drought. Opisthopappus taihangensis (ling) shih is a typical drought-tolerant perennial plant species endemically distributed across the Taihang Mountains in China, but the underlying mechanism for drought tolerance of this particular species remains elusive. RESULTS: To mimic natural drought stress, O. taihangensis plants were treated with two different concentrations (25% and 5%) of polyethylene glycol (PEG6000), which represent the H group (high salinity) and the L group (low salinity), respectively. The physiological characteristics of these two groups of plants, including relative water content maintenance (RWC), proline content and chlorophyll content were assessed and compared with plants in the control group (CK), which had normal irrigation. There was not a significant difference in RWC when comparing plants in the L group with the control group. Proline was accumulated to a higher level, and chlorophyll content was decreased slightly in plants under low drought stress. In plants from the H group, a lower RWC was observed. Proline was accumulated to an even higher level when compared with plants from the L group, and chlorophyll content was further reduced in plants under high drought stress. Transcriptomic analysis was carried out to look for genes that are differentially expressed (DEGs) in O. taihangensis plants coping adaptively with the two levels of drought stress. A total of 23,056 genes are differentially expressed between CK and L, among which 12,180 genes are up-regulated and 10,876 genes are down-regulated. Between H and L, 6182 genes are up-regulated and 1850 genes are down-regulated, which gives a total of 8032 genes. The highest number of genes, that are differentially expressed, was obtained when a comparison was made between CK and H. A total of 43,074 genes were found to be differentially expressed with 26,977 genes up-regulated and 16,097 genes down-regulated. Further analysis of these genes suggests that many of the up-regulated genes are enriched in pathways involved in amino acid metabolism. Besides, 39 transcription factors (TFs) were found to be continuously up-regulated with the increase of drought stress level. CONCLUSION: Taken together, the results indicate that O. taihangensis plants are able to live adaptively under drought stress by responding physiologically and regulating the expression of a substantial number of drought-responsive genes and TFs to avoid adverse effects.