1. 领域背景与文献引入
核心信息段:文献英文标题:HTT loss-of-function contributes to RNA deregulation in developing Huntington’s disease neurons;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:神经退行性疾病(亨廷顿舞蹈症)。
亨廷顿舞蹈症(HD)是一种常染色体显性遗传的神经退行性疾病,1993年研究人员首次发现其致病机制为HTT基因中CAG三核苷酸重复序列扩增,导致编码的亨廷顿蛋白(HTT)出现异常长聚谷氨酰胺(polyQ)链,这一发现成为领域发展的关键节点。此后的研究围绕突变HTT(mutHTT)的获得性功能(GoF)毒性展开,证实mutHTT会形成聚集物、破坏细胞内信号通路,最终导致纹状体中型多棘神经元(MSN)死亡,这一机制长期被认为是HD的核心致病原因。近年来,领域内逐渐认识到野生型HTT(wtHTT)的功能缺失(LoF)也参与HD发病,小鼠模型研究显示,完全敲除HTT基因会导致胚胎致死,仅保留50%wtHTT表达的小鼠会出现皮层和纹状体畸形,提示wtHTT在神经发育和神经元存活中具有关键作用。当前研究热点聚焦于HD的早期分子改变,越来越多的证据表明HD是一种神经发育疾病,成年期的神经元退行性变源于发育阶段的转录失调,但目前对于早期转录失调的具体RNA调控网络,以及wtHTT功能缺失在其中的贡献仍不明确,这一核心问题限制了HD早期诊断标志物和靶向治疗策略的开发。
针对这一研究空白,本研究采用遗传背景一致的人类神经干细胞模型,同时对比HD细胞、HTT完全敲除(KO)细胞和对照细胞的转录组与小RNA组变化,旨在揭示HD早期神经发育阶段的RNA失调网络,明确wtHTT功能缺失在早期转录失调中的核心作用,为HD的早期干预提供新的分子靶点和理论依据。
2. 文献综述解析
核心信息段:作者在综述部分以HD的致病机制为核心分类维度,将现有研究分为mutHTT获得性功能毒性和wtHTT功能缺失两大方向,同时结合研究模型(小鼠模型、人类干细胞模型)的差异,系统梳理了领域内的研究进展与未解决问题。
现有研究中,mutHTT获得性功能毒性是经典的致病机制,大量小鼠模型和人类病理样本研究证实,mutHTT会通过与转录因子、染色质调控因子等相互作用,破坏细胞内稳态,导致神经元功能障碍和死亡,这一机制的研究较为成熟,为HD的靶向治疗提供了基础。wtHTT功能缺失的研究则主要基于小鼠模型,完全敲除HTT基因的小鼠会在胚胎期死亡,部分缺失wtHTT的小鼠会出现脑结构畸形、运动异常等神经表型,提示wtHTT在神经发育和维持神经元功能中不可或缺。人类干细胞模型的研究则进一步发现,HD患者来源的诱导多能干细胞(iPSC)在分化为神经元的过程中,早期就会出现转录失调,这一发现将HD的发病时间线提前至神经发育阶段。现有研究的技术方法各有优势,小鼠模型能在整体动物水平研究HTT的功能,人类干细胞模型则更贴近人类病理特征,但也存在明显局限性:小鼠与人类存在物种差异,其研究结果难以直接外推至人类;多数研究未同时对比HD细胞和HTT完全敲除细胞,无法区分mutHTT获得性功能和wtHTT功能缺失对早期转录失调的贡献;针对HD早期神经发育阶段的RNA调控网络研究仍不系统,缺乏对mRNA和miRNA协同调控的分析。
通过对比现有研究的未解决问题,本研究的创新价值凸显:首次使用遗传背景完全一致的人类神经干细胞模型(HD、HTT-KO、对照),排除了遗传异质性的干扰;同时分析mRNA和miRNA的表达变化,构建了完整的RNA调控网络;通过拯救实验直接验证了wtHTT功能缺失对转录失调的贡献,明确了其在HD早期发病中的核心作用,弥补了领域内对两种致病机制区分研究的不足。
3. 研究思路总结与详细解析
核心信息段:本研究的整体研究目标是揭示HD早期神经发育阶段转录失调的RNA调控网络,明确wtHTT功能缺失在其中的核心作用;核心科学问题是HD中mutHTT获得性功能和wtHTT功能缺失哪个是早期转录失调的主要驱动因素;技术路线遵循“模型构建→组学分析→验证实验→机制解析→网络构建”的闭环逻辑,通过同基因细胞模型的对比研究,逐步明确HTT功能缺失的致病作用。
3.1 同基因神经细胞模型构建与验证
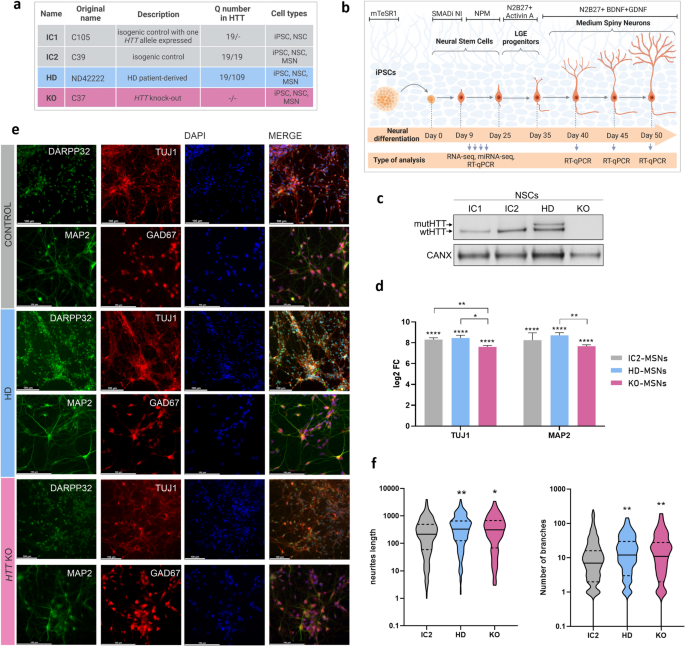
实验目的:构建遗传背景一致的HD、HTT-KO和对照神经干细胞(NSC)及中型多棘神经元(MSN)样细胞,为后续组学分析和功能实验提供标准化模型。方法细节:使用CRISPR-Cas9技术构建的同基因iPSC系(HD、IC1、IC2、KO),通过STEMdiff SMADi神经诱导试剂盒将iPSC分化为NSC,再通过已发表的方案将NSC分化为MSN样细胞;通过蛋白质免疫印迹(WB)检测HTT蛋白表达水平,通过实时荧光定量PCR(RT-qPCR)检测神经元标记物TUJ1、MAP2的表达,通过免疫细胞化学(ICC)检测神经元标记物(MAP2、TUJ1)和纹状体特异性标记物(GAD67、DARPP32),并使用Cellprofiler软件定量分析神经突长度和分支数。结果解读:WB结果显示,KO-NSC中无HTT蛋白表达,IC1-NSC中wtHTT表达水平显著降低,符合预期的基因编辑结果;RT-qPCR结果显示,MSN样细胞中TUJ1和MAP2的表达较iPSC升高150倍以上,证实神经元分化成功,其中KO-MSN中TUJ1的表达显著低于其他组(n=3,P<0.01),提示HTT缺失影响神经元成熟;ICC染色结果显示,所有组的MSN样细胞均表达神经元和纹状体标记物,证实细胞类型的正确性;定量分析显示,HD-MSN和KO-MSN的神经突长度和分支数较对照组显著增加(IC2组n=523,HD组n=352,KO组n=198,P<0.0001),提示早期神经发育阶段的形态学改变。
产品关联:实验所用关键产品:STEMCELL Technologies的STEMdiff SMADi Neural Induction Kit,Thermo Fisher Scientific的Accutase、4%多聚甲醛,Leica的DMI6000显微镜,Cellprofiler 4.2.8软件等。
3.2 转录组与小RNA组测序分析
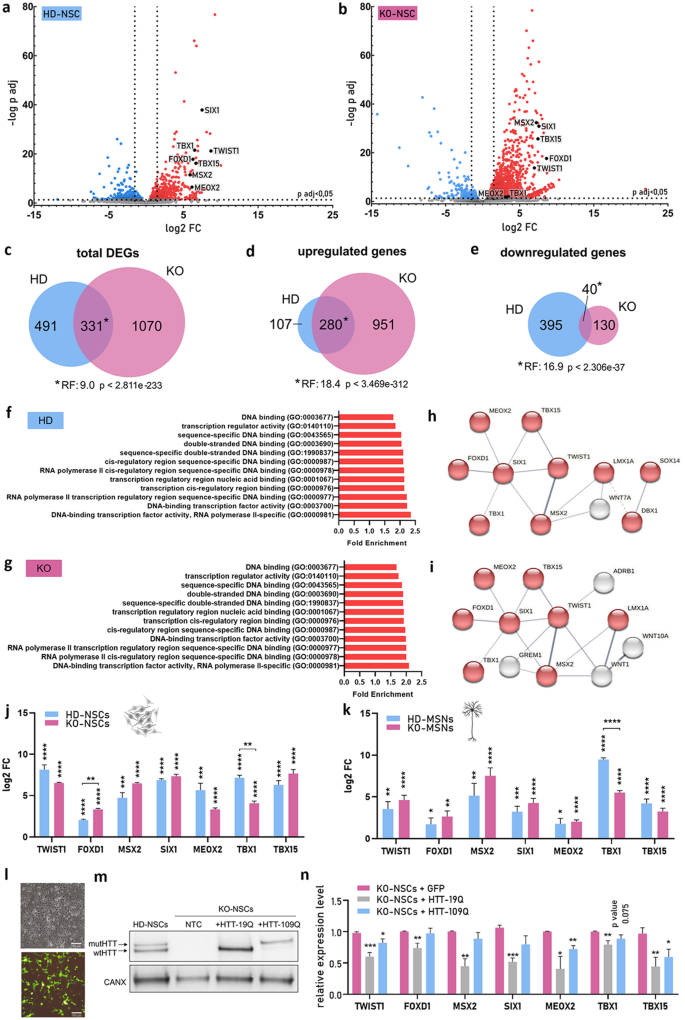
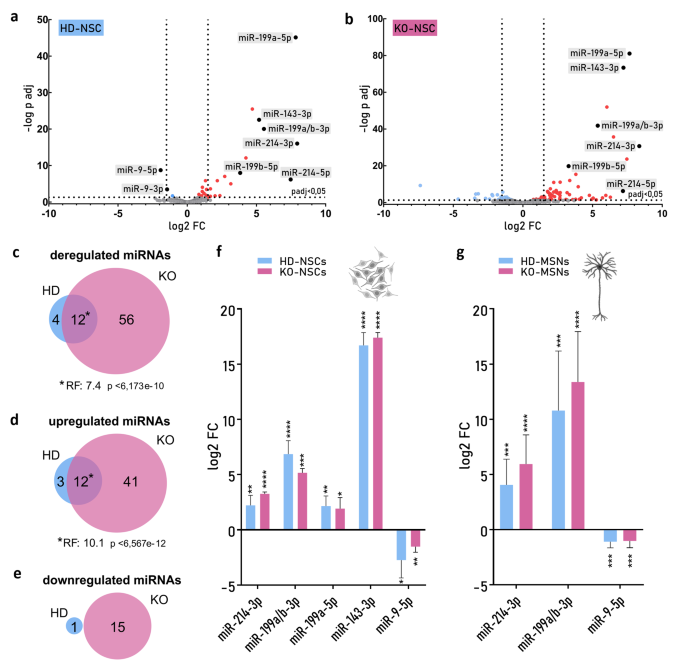
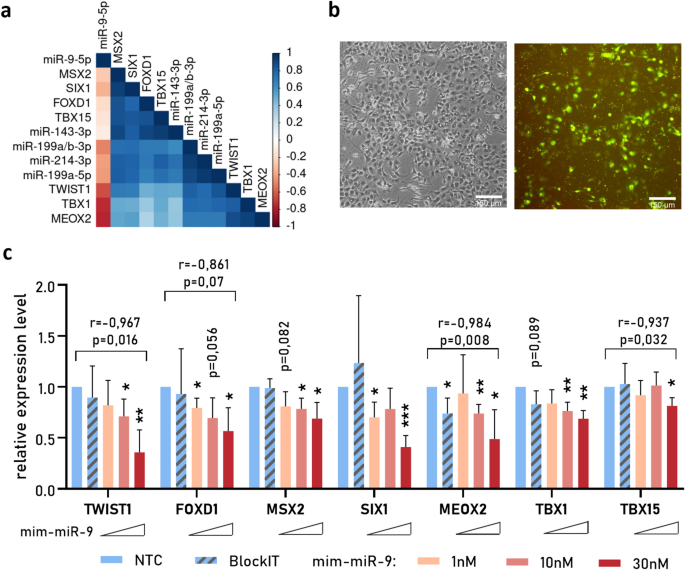
实验目的:分析HD、KO和对照NSC的mRNA和miRNA表达差异,筛选出与HTT功能缺失相关的失调RNA网络。方法细节:从3个生物学重复的对照NSC和4个生物学重复的HD、KO NSC中提取总RNA,使用Illumina NovaSeq 6000系统进行RNA-seq(总RNA测序深度为50 MR/样本,小RNA测序深度为10 MR/样本);通过生物信息学分析筛选差异表达基因(DEG,|log2FC|>1.5,padj<0.05)和差异表达miRNA,使用PANTHER进行基因本体(GO)富集分析,使用STRING构建蛋白质相互作用网络,通过Spearman相关分析研究基因与miRNA的表达相关性。结果解读:RNA-seq结果显示,KO-NSC中共有1401个差异表达基因,HD-NSC中共有822个差异表达基因,两者有331个重叠差异基因,重叠率具有统计学显著性(表示因子RF=9,P<0.05),重叠的上调基因显著富集于DNA结合和转录调控相关的转录因子;小RNA-seq结果显示,KO-NSC中有68个差异表达miRNA,HD-NSC中有16个差异表达miRNA,两者有12个重叠差异miRNA(RF=7.4,P<0.05),包括上调的miR-214-3p、miR-199a/b-3p和下调的miR-9-5p等。
产品关联:实验所用关键产品:Zymo Research的Direct-zol RNA Microprep试剂盒,Agilent的RNA Pico 6000 kit和Bioanalyzer 2100,Illumina的NovaSeq 6000系统,PANTHER v.17、STRING v11.5生物信息学工具等。
3.3 关键转录因子的验证与拯救实验
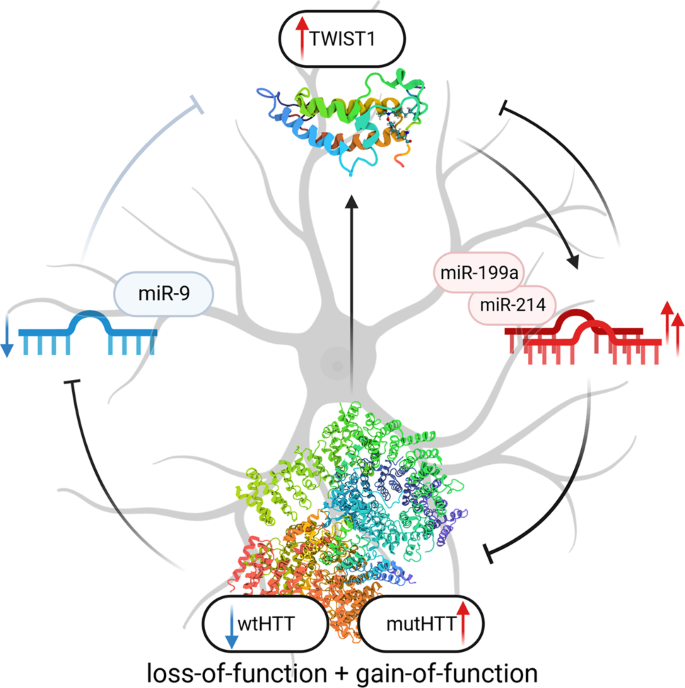
实验目的:验证RNA-seq筛选出的失调转录因子,并确认其表达变化与wtHTT功能缺失的直接关联。方法细节:通过RT-qPCR检测7个上调的转录因子(TWIST1、SIX1、TBX1、TBX15、MSX2、MEOX2、FOXD1)在HD、KO和对照NSC及MSN中的表达水平;通过Neon电转染系统将表达wtHTT(19Q)或mutHTT(109Q)的质粒转入KO-NSC,48小时后通过WB验证HTT蛋白表达,通过RT-qPCR检测转录因子的表达变化。结果解读:RT-qPCR结果显示,7个转录因子在HD和KO的NSC及MSN中均显著上调,与RNA-seq结果一致(P<0.05);拯救实验显示,转入wtHTT质粒后,KO-NSC中所有7个转录因子的表达水平显著下调40%-60%(wtHTT组n=4,P<0.05),转入mutHTT质粒后,仅TWIST1、MEOX2、TBX15的表达水平下调20%-40%(mutHTT组n=5,P<0.05),提示wtHTT功能缺失是这些转录因子上调的主要原因,mutHTT的拯救作用较弱可能与其功能异常有关。
产品关联:实验所用关键产品:Addgene的HTT-19Q(货号#111741)和HTT-109Q(货号#111730)质粒,Invitrogen的Neon Transfection System,Bio-Rad的CFX Connect Real-Time System、SsoAdvanced Universal SYBR Green Supermix等。
3.4 关键miRNA的验证与功能实验
实验目的:验证RNA-seq筛选出的失调miRNA,并分析其对靶转录因子的调控作用。方法细节:通过RT-qPCR检测miR-214-3p、miR-199a/b-3p、miR-9-5p在HD、KO和对照NSC及MSN中的表达水平;通过Lipofectamine 2000将不同浓度的miR-9 mimic转入HD-NSC,24小时后通过RT-qPCR检测转录因子的表达变化。结果解读:RT-qPCR结果显示,miR-214-3p、miR-199a/b-3p在HD和KO的NSC及MSN中显著上调,miR-9-5p显著下调,与RNA-seq结果一致(P<0.05);miR-9 mimic转染实验显示,HD-NSC中7个转录因子的表达水平呈剂量依赖性下调,30 nM mimic处理后,转录因子表达降低35%-80%(n=3,P<0.05),其中TWIST1、MEOX2、TBX15的表达与miR-9浓度呈负相关,证实miR-9对这些转录因子具有负调控作用。
产品关联:实验所用关键产品:Thermo Fisher的Lipofectamine 2000、miRVana miR-9 mimic(货号#4464066),Applied Biosystems的TaqMan Advanced miRNA cDNA Synthesis Kit、TaqMan Advanced miRNA Assays等。
4. Biomarker研究及发现成果
核心信息段:本研究中涉及的Biomarker包括转录因子(TWIST1、SIX1、TBX1、TBX15、MSX2、MEOX2、FOXD1)和miRNA(miR-214-3p、miR-199a/b-3p、miR-9-5p),筛选与验证逻辑为“组学筛选→细胞水平验证→功能实验确认”,通过同基因细胞模型的对比,明确了这些Biomarker与HTT功能缺失的关联。
Biomarker定位:这些Biomarker属于HD早期神经发育阶段的转录调控类Biomarker,筛选逻辑为通过RNA-seq在HD和HTT-KO细胞中筛选出重叠失调的分子,排除了mutHTT获得性功能特异性的变化,聚焦于HTT功能缺失相关的失调网络;验证逻辑为通过RT-qPCR在NSC和MSN两种细胞类型中验证表达变化,通过拯救实验和miR mimic实验确认其与HTT功能的调控关系,形成了完整的证据链。
研究过程详述:这些Biomarker来源于人类iPSC衍生的神经干细胞和MSN样细胞,模拟了HD早期神经发育阶段的病理状态;验证方法包括RT-qPCR定量表达水平,拯救实验验证wtHTT对转录因子的调控作用,miR mimic实验验证miRNA对转录因子的调控作用;特异性方面,这些分子在HD和HTT-KO细胞中均显著失调,与对照细胞存在明显差异(|log2FC|>1.5,padj<0.05),RT-qPCR验证结果与组学数据一致,具有统计学显著性(P<0.05);敏感性方面,组学分析和验证实验均能稳定检测到这些分子的表达变化,提示其具有作为早期Biomarker的潜力。
核心成果提炼:本研究首次明确了由这些转录因子和miRNA构成的前馈调控网络,miR-9负调控TWIST1等转录因子的表达,TWIST1调控miR-214/199a簇的表达,而miR-214又反向调控HTT的表达,形成了一个相互作用的调控环;首次证实HD早期转录失调主要由wtHTT功能缺失驱动,70%以上的HD上调基因与HTT-KO细胞重叠;其中TWIST1的上调与HD患者脑组织中的变化一致,提示其可作为HD早期诊断的潜在Biomarker,不过原文未提供该分子在临床样本中的风险比、敏感性等具体数据,需进一步临床验证。此外,miR-9、miR-214等miRNA也可作为HD早期干预的潜在靶点,为HD的精准治疗提供了新方向。