1. 领域背景与文献引入
文献英文标题:The suppression of HSPA8 attenuates NLRP3 ubiquitination through SKP2 to promote pyroptosis in sepsis-induced lung injury;发表期刊:Cell Bioscience;影响因子:未公开;研究领域:脓毒症诱导急性肺损伤的分子机制(重症医学与分子免疫学交叉领域)
领域共识:脓毒症是重症监护病房(ICU)患者的主要死亡原因之一,急性肺损伤(ALI)是其最常见且进展最快的器官损伤类型,病死率可高达40%。当前临床主要依赖机械通气、抗炎药物及液体保守治疗等手段,但治疗效果仍不理想,亟需明确脓毒症诱导ALI的核心分子机制以开发靶向治疗策略。近年研究证实,NOD样受体热蛋白结构域相关蛋白3(NLRP3)炎症小体介导的焦亡是脓毒症进展的关键环节,但肺泡上皮细胞中NLRP3炎症小体的具体激活调控机制尚未明确,这一空白限制了针对性治疗靶点的开发。
针对上述领域空白,本研究聚焦热休克蛋白家族A成员8(HSPA8)在脓毒症诱导ALI中的功能与调控机制,旨在揭示HSPA8通过E3泛素连接酶S期激酶相关蛋白2(SKP2)调控NLRP3泛素化水平的分子通路,为脓毒症诱导ALI的治疗提供新的潜在靶点与理论依据。
2. 文献综述解析
作者以“临床问题-分子机制-研究空白”的逻辑构建综述体系,先从脓毒症ALI的临床困境切入,逐层聚焦至焦亡、NLRP3炎症小体、HSPA8及SKP2的研究进展,最终明确当前领域的核心未解决问题。
现有研究已证实NLRP3炎症小体激活是介导脓毒症多器官损伤的核心分子事件,其通过切割半胱天冬酶-1(Caspase-1)和Gasdermin D(GSDMD)诱导焦亡,释放促炎因子放大炎症反应;HSPA8作为组成型表达的分子伴侣,参与蛋白折叠、应激反应及泛素化降解调控,在多种疾病中发挥保护或损伤作用;SKP2作为SCF泛素连接酶复合物的核心组分,介导底物蛋白的K48位泛素化降解,参与细胞周期调控与肿瘤发生。但现有研究未将HSPA8、SKP2与NLRP3炎症小体的调控关联起来,且缺乏在肺泡上皮细胞这一脓毒症ALI关键效应细胞中的机制验证,无法为临床治疗提供直接的分子靶点。
本研究的创新价值在于首次建立了HSPA8-SKP2-NLRP3的调控通路,明确了HSPA8通过稳定SKP2蛋白水平,促进NLRP3的泛素化降解,进而抑制肺泡上皮细胞焦亡的分子机制,填补了脓毒症诱导ALI中NLRP3炎症小体上游调控机制的空白,为开发靶向干预策略提供了新的理论基础。
3. 研究思路总结与详细解析
本研究以“明确HSPA8在脓毒症诱导ALI中的功能→解析其介导的细胞死亡类型→揭示下游调控通路→验证中间分子的作用”为核心技术路线,形成“假设-实验验证-机制解析-体内验证”的完整研究闭环,最终明确HSPA8通过SKP2调控NLRP3泛素化的分子机制。
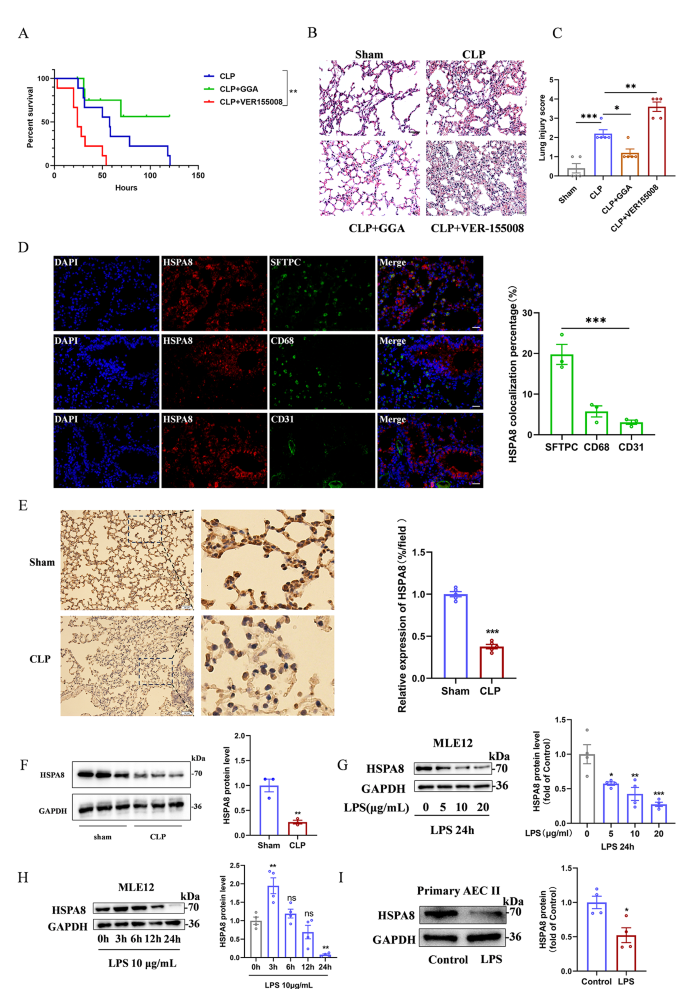
3.1 体内外模型构建与HSPA8表达验证
实验目的:确认HSPA8在脓毒症诱导ALI中的表达变化及对疾病进展的影响。
方法细节:构建盲肠结扎穿刺(CLP)小鼠脓毒症模型,将小鼠分为假手术组、CLP组、CLP+HSPA8激动剂香叶基香叶基丙酮(GGA)组、CLP+HSPA8抑制剂VER155008组,通过存活率统计、肺组织病理染色评估疾病严重程度;细胞层面采用脂多糖(LPS)联合三磷酸腺苷(ATP)处理小鼠肺泡上皮细胞(MLE12)及原代Ⅱ型肺泡上皮细胞(AECⅡ),通过免疫组化、免疫荧光、蛋白质免疫印迹(Western blot)检测HSPA8的表达水平。
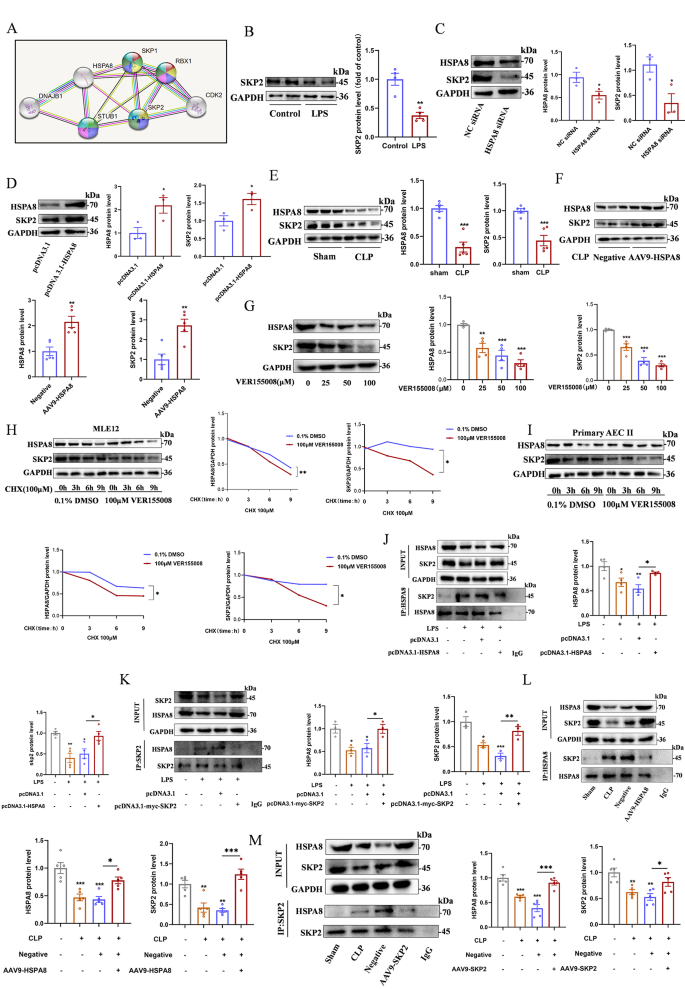
结果解读:CLP小鼠肺组织中HSPA8蛋白水平显著下调(n=5,P<0.01),LPS处理后MLE12细胞中HSPA8表达随浓度升高和时间延长呈依赖性降低(n=4,P<0.001);GGA预处理可显著提高CLP小鼠的存活率(n=9,P<0.05),而VER155008处理则加重肺组织病理损伤,肺损伤评分达3.2±0.3(n=5,P<0.001)。免疫荧光结果显示HSPA8在CLP小鼠的AECⅡ中广泛表达,提示其可能通过调控肺泡上皮细胞功能参与疾病进展。
产品关联:文献未提及具体实验产品,领域常规使用Western blot相关一抗/二抗、细胞培养试剂、动物造模手术器械等。
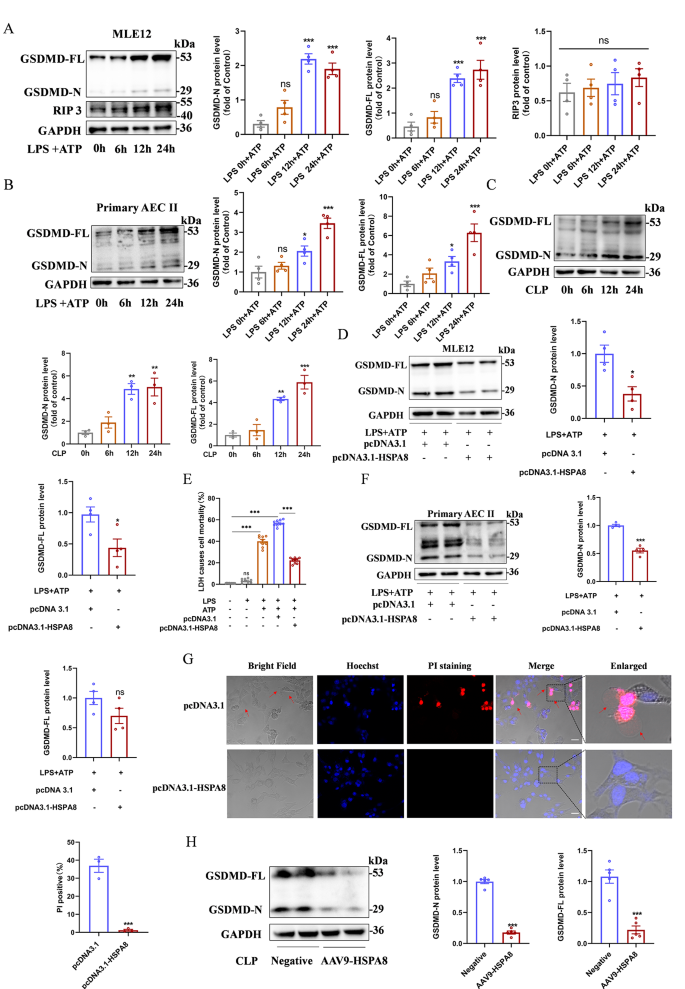
3.2 HSPA8调控肺泡上皮细胞焦亡的功能验证
实验目的:明确HSPA8是否通过调控焦亡介导脓毒症诱导的肺泡上皮细胞损伤。
方法细节:采用Hoechst33342/碘化丙啶(PI)双染色、乳酸脱氢酶(LDH)释放实验检测细胞死亡类型;通过质粒转染过表达HSPA8,或用小干扰RNA(siRNA)敲低HSPA8,结合Western blot检测焦亡标志物GSDMD-N端(GSDMD-N)的表达水平;在动物层面通过腺相关病毒9(AAV9)介导HSPA8过表达,验证其对肺组织焦亡的调控作用。
结果解读:LPS+ATP处理后MLE12细胞中GSDMD-N表达显著升高(n=4,P<0.01),而受体相互作用蛋白3(RIP3)表达无明显变化,提示细胞死亡类型为焦亡;过表达HSPA8可显著降低LPS+ATP诱导的GSDMD-N表达及LDH释放(n=9,P<0.001);AAV9-HSPA8处理可显著减少CLP小鼠肺组织中GSDMD-N的蛋白水平(n=5,P<0.01),证实HSPA8可通过抑制焦亡减轻肺泡上皮细胞损伤。
产品关联:文献未提及具体实验产品,领域常规使用LDH检测试剂盒、GSDMD特异性抗体、质粒转染试剂等。
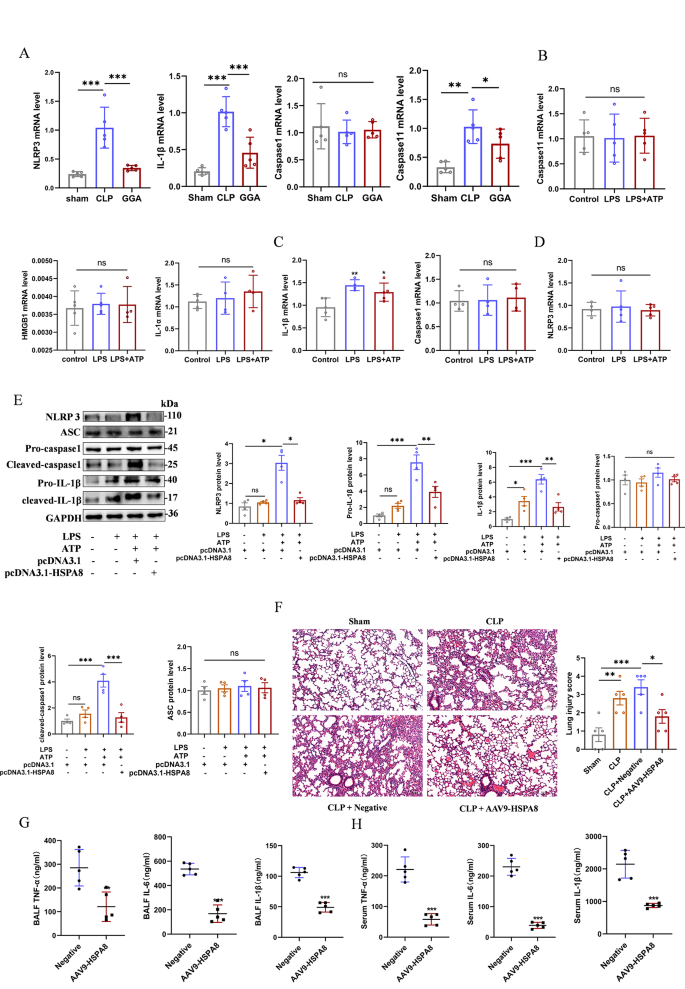
3.3 HSPA8对NLRP3炎症小体的调控机制解析
实验目的:揭示HSPA8抑制焦亡的下游分子通路,明确其对NLRP3炎症小体的调控作用。
方法细节:通过实时荧光定量PCR(qRT-PCR)检测NLRP3炎症小体相关分子的mRNA表达,Western blot检测蛋白水平;采用免疫共沉淀(Co-IP)检测NLRP3的泛素化修饰水平;在动物层面检测肺泡灌洗液及血清中促炎因子的释放水平。
结果解读:CLP小鼠肺组织中NLRP3、白细胞介素-1β(IL-1β)的mRNA水平显著升高(n=5,P<0.01),GGA预处理可显著降低上述分子的表达;过表达HSPA8可抑制LPS+ATP诱导的NLRP3、活化型Caspase-1及活化型IL-1β的蛋白表达(n=5,P<0.01);Co-IP结果显示HSPA8过表达可显著增加NLRP3的泛素化水平(n=3,P<0.05),减少其蛋白积累;AAV9-HSPA8处理可显著降低CLP小鼠肺泡灌洗液及血清中肿瘤坏死因子-α(TNF-α)、IL-6、IL-1β的释放水平(n=5,P<0.01),证实HSPA8通过促进NLRP3泛素化降解抑制炎症小体激活。
产品关联:文献未提及具体实验产品,领域常规使用qRT-PCR试剂盒、Co-IP相关磁珠与抗体、酶联免疫吸附试验(ELISA)试剂盒等。
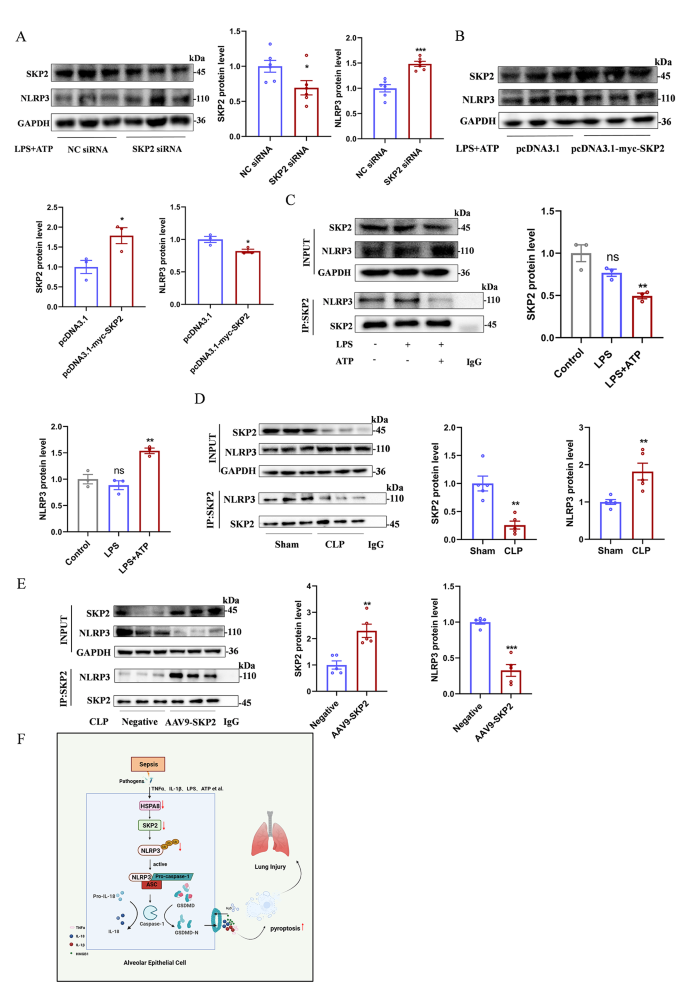
3.4 SKP2作为中间调控分子的功能验证
实验目的:明确HSPA8调控NLRP3泛素化的中间分子,验证HSPA8-SKP2-NLRP3通路的完整性。
方法细节:通过STRING数据库预测HSPA8与SKP2的相互作用,采用Co-IP验证两者的结合关系;通过siRNA敲低或质粒过表达HSPA8,检测SKP2的蛋白水平与稳定性;过表达SKP2后检测NLRP3泛素化水平及炎症小体激活情况;在动物层面通过AAV9-SKP2验证其对脓毒症诱导ALI的治疗作用。
结果解读:Co-IP结果证实HSPA8与SKP2存在直接相互作用,敲低HSPA8可显著降低SKP2的蛋白水平(n=4,P<0.01),并缩短其蛋白半衰期(n=4,P<0.05);过表达SKP2可显著增加NLRP3的泛素化水平(n=3,P<0.05),抑制LPS+ATP诱导的GSDMD-N表达及LDH释放(n=10,P<0.001);AAV9-SKP2处理可显著减轻CLP小鼠的肺组织病理损伤,降低促炎因子释放水平(n=5,P<0.01),证实SKP2是HSPA8调控NLRP3泛素化的关键中间分子。
产品关联:文献未提及具体实验产品,领域常规使用siRNA转染试剂、AAV载体构建服务、泛素化检测抗体等。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究的核心功能Biomarker为HSPA8,其筛选与验证遵循“临床模型发现-体内外功能验证-分子机制解析”的完整逻辑链:先通过CLP小鼠模型发现HSPA8表达与脓毒症诱导ALI的严重程度负相关,再通过细胞与动物实验验证其抑制焦亡的功能,最终解析其通过SKP2调控NLRP3泛素化的分子机制。
研究过程详述
HSPA8的样本来源为脓毒症小鼠的肺组织及肺泡上皮细胞,验证方法包括免疫组化、Western blot、qRT-PCR等,功能验证通过HSPA8激动剂/抑制剂处理、基因过表达及敲低等手段完成。特异性与敏感性分析显示,CLP小鼠肺组织中HSPA8表达水平与肺损伤评分呈显著负相关(VER155008组肺损伤评分3.2±0.3,n=5,P<0.001),GGA诱导HSPA8过表达可使CLP小鼠的存活率提高约40%(n=9,P<0.05),证实HSPA8具有作为脓毒症诱导ALI病情评估指标及治疗靶点的潜力。
核心成果提炼
本研究首次揭示HSPA8可作为脓毒症诱导ALI的潜在治疗靶点,其通过与SKP2结合维持蛋白稳定性,进而促进NLRP3的泛素化降解,最终抑制NLRP3炎症小体激活与肺泡上皮细胞焦亡。统计学结果显示,过表达HSPA8可使LPS+ATP处理的MLE12细胞LDH释放水平降低42%(n=9,P<0.001),使CLP小鼠血清中IL-1β水平降低38%(n=5,P<0.01)。该成果不仅完善了脓毒症诱导ALI的分子调控网络,更为开发靶向HSPA8或SKP2的新型治疗策略提供了直接的实验依据。