1. 领域背景与文献引入
文献英文标题:Transcriptome-wide association studies associated with Crohn’s disease: challenges and perspectives;发表期刊:Cell Bioscience;影响因子:未公开;研究领域:炎症性肠病(克罗恩病)的转录组学与遗传学交叉研究
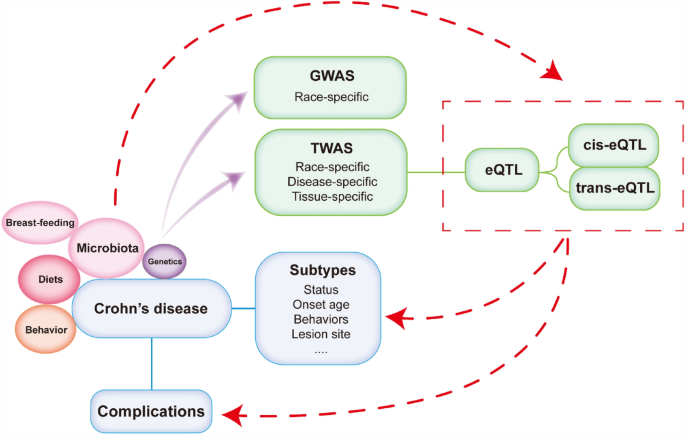
炎症性肠病是一类慢性复发性肠道免疫紊乱疾病,其中克罗恩病(CD)为终身进展性亚型,可累及全胃肠道,临床表型具有高度异质性,蒙特利尔分类从发病年龄、病变部位、疾病行为等维度对其进行分层。过去二十余年,全基因组关联研究(GWAS)取得突破性进展,2005至2022年间NHGRI-EBI GWAS数据库收录了约40万个与人类性状相关的单核苷酸多态性(SNP),针对CD的研究已发现超过240个炎症性肠病易感基因或位点,其中37个为CD特异性位点。但GWAS存在显著局限性,约90%的关键信号位于非编码区,无法直接解释其致病功能。转录组全关联研究(TWAS)作为整合GWAS与表达数量性状位点(eQTL)数据的生物信息学方法,可通过预测基因表达水平关联疾病风险,为解析非编码区SNP功能、筛选候选致病基因提供新途径。然而,目前CD的TWAS研究结果重复性较低,研究设计异质性对结果的影响尚不明确,亟需系统梳理现有研究以总结规律、优化策略。本文旨在综述已发表的7项CD TWAS研究,分析其研究设计、方法学差异及易感基因特征,探讨结果重复性低的原因并提出未来方向。
2. 文献综述解析
本文对领域内7项CD TWAS研究进行系统综述,以研究设计要素(人群、组织类型、eQTL选择、分析方法)为分类维度,总结现有研究成果与局限性,明确CD TWAS研究的核心挑战与未来方向。
现有研究可按人群分为东亚(日本、韩国)与欧美(美国、英国)队列,按组织类型分为单组织(回肠、结肠、血液)与跨组织研究,按分析方法分为基于PrediXcan/Fusion的传统方法与基于UTMOST的跨组织预测方法。关键结论包括:不同组织中筛选出多个重叠的CD易感基因,如回肠中的ATG16L1、ERAP2,结肠中的ERAP2、IL23R,血液中的NOD2、TNFSF15;这些基因分别参与肠道屏障功能、抗原提呈、免疫调节等关键通路;GO功能富集分析显示,回肠易感基因富集于脂质代谢通路,结肠易感基因富集于囊泡膜相关细胞组分,血液易感基因富集于生物刺激应答的免疫调节通路。技术方法层面,TWAS的优势在于整合GWAS与eQTL数据,弥补GWAS无法解释非编码区SNP功能的不足,实现对难以获取的肠道病变组织的基因表达与疾病关联分析;但现有研究存在明显局限性,包括结果重复性低,不同研究间易感基因重叠率低,部分研究样本量较小,疾病亚型信息缺失,eQTL选择的种族与组织特异性导致结果差异显著。
与现有零散的TWAS研究相比,本文首次系统整合7项CD TWAS研究结果,从研究设计全流程(基因型数据选择、eQTL数据匹配、分析方法选择、组织类型差异)解析结果异质性来源,通过GO功能富集分析揭示组织特异性致病通路,明确回肠脂质代谢与结肠细胞外囊泡在CD发病中的潜在作用;同时深入探讨现有研究重复性低的核心原因,包括CD亚型信息不足、eQTL设计的种族与组织异质性、分析方法多样性等,并提出针对性未来研究策略,填补了领域内对CD TWAS研究系统总结的空白。
3. 研究思路总结与详细解析
本文研究目标是系统梳理CD TWAS研究现状,分析研究设计异质性对结果的影响,总结易感基因与功能通路,探讨结果重复性低的原因并提出未来方向;核心科学问题是CD TWAS研究结果重复性低的关键影响因素,以及如何优化研究设计以提高结果可靠性;技术路线遵循“文献纳入→分层分析→基因整合→功能富集→问题分析→策略提出”的闭环逻辑。
3.1 文献检索与纳入分析
实验目的是筛选所有已发表的CD相关TWAS研究,明确研究基本特征;方法细节:通过学术数据库检索获取相关文献,最终纳入7项符合标准的研究,提取每项研究的人群特征、组织类型、eQTL数据来源、分析方法、易感基因结果等核心信息;结果解读:纳入的7项研究中,4项为单组织TWAS研究,分别针对日本、韩国、美国、英国人群,其中日本研究同时使用跨组织eQTL数据库;3项为跨组织与多人群研究;研究人群涵盖东亚与欧美,组织类型包括回肠、结肠、肠道免疫细胞、血液免疫细胞、全血;其中Dai等的研究使用了英国IBD-BIOM队列的24例CD患者与23例健康对照(n=47),Gettler等使用了RISK队列的儿童CD研究样本;产品关联:文献未提及具体实验产品,领域常规使用文献管理软件(如EndNote)、学术数据库检索工具(如PubMed、Web of Science)。
3.2 研究设计要素分层分析
实验目的是解析CD TWAS研究结果异质性的来源,明确各设计要素对结果的影响;方法细节:从基因型数据、eQTL数据、分析方法、组织类型四个核心维度对7项研究进行分层对比分析,其中基因型数据关注人群种族一致性与样本量,eQTL数据关注组织特异性、种族匹配性与数据库选择,分析方法关注不同工具的差异,组织类型关注不同肠道部位与免疫相关组织的结果差异;结果解读:基因型数据的种族一致性显著影响结果准确性,如日本研究使用GTEx数据库(欧美人群)仅筛选出1个全血易感基因,而韩国研究使用本土eQTL数据库筛选出21个;eQTL的组织特异性导致不同组织的易感基因差异显著,回肠与结肠的功能通路完全不同;不同分析方法筛选的基因重叠率低,如UTMOST与MetaXcan筛选的基因几乎无重叠;
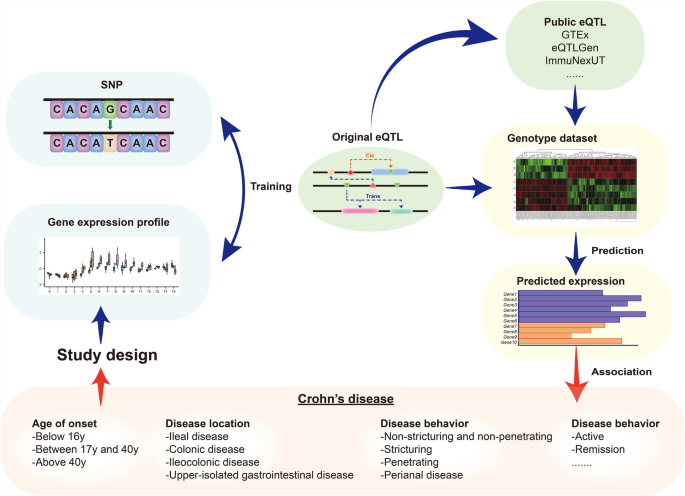
产品关联:文献未提及具体实验产品,领域常规使用生物信息学分析工具(如R语言、Python)、TWAS专用分析软件(如PrediXcan、Fusion)。
3.3 易感基因整合与功能富集分析
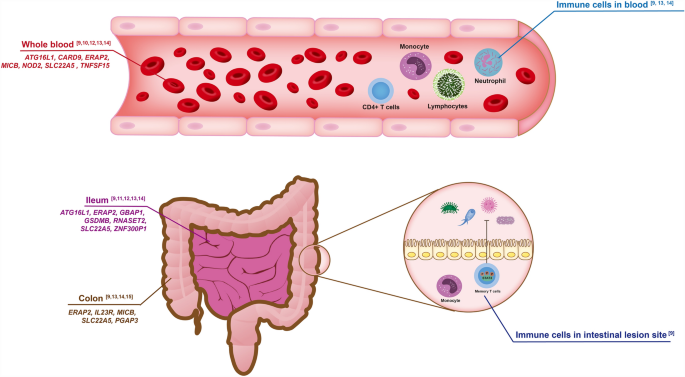
实验目的是整合不同组织中的CD易感基因,解析其功能通路特征;方法细节:提取7项研究中各组织的所有易感基因,使用R语言的clusterProfiler与pathview包进行GO功能富集分析,由于缺乏基因表达的差异倍数,随机分配foldchange值为1或-1进行粗略富集;结果解读:回肠易感基因富集于脂质代谢相关生物过程,结肠易感基因富集于囊泡膜等细胞组分,血液易感基因富集于免疫应答与生物刺激调节通路;不同组织中存在多个重叠易感基因,如ERAP2在回肠、结肠、血液中均被筛选到,ATG16L1在回肠与血液中被重复验证;
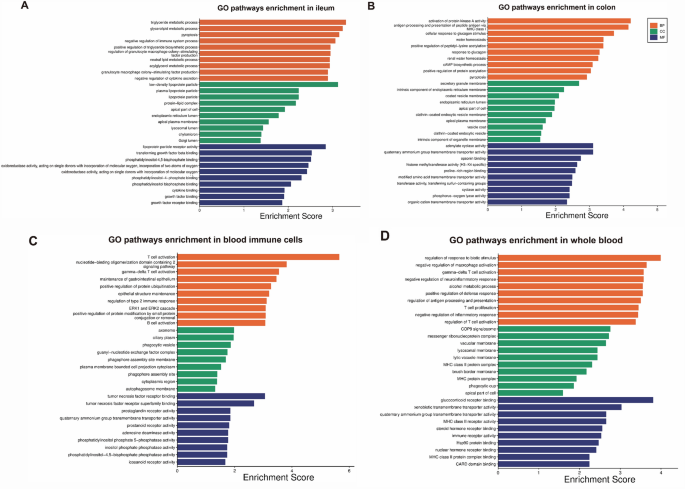
产品关联:文献未提及具体实验产品,领域常规使用R语言的生物信息学分析包(如clusterProfiler、pathview)、基因功能注释数据库(如GO、KEGG)。
3.4 重复性问题分析与未来方向提出
实验目的是探讨CD TWAS研究结果重复性低的原因,提出优化研究设计的策略;方法细节:对比不同研究的结果差异,结合CD的疾病特征与领域研究现状,分析重复性低的核心原因,并提出针对性建议;结果解读:重复性低的原因包括CD亚型信息缺失(如发病年龄、病变部位、疾病活动度)、eQTL设计的种族与组织异质性、分析方法的快速更新、样本量不足等;未来研究需构建大样本多中心的GWAS数据库,包含详细的疾病亚型信息;建立分类更全面的eQTL数据库,涵盖不同种族、组织、疾病状态;结合单细胞转录组等新技术开展TWAS研究;
产品关联:文献未提及具体实验产品,领域常规使用单细胞转录组测序平台(如10x Genomics)、多组学整合分析平台。
4. Biomarker研究及发现成果解析
本文中涉及的Biomarker为CD的组织特异性易感基因,通过TWAS整合GWAS与eQTL数据在不同组织中筛选,并通过多个独立研究的重叠验证确定其可靠性;这些Biomarker涵盖回肠、结肠、血液等多个组织,参与肠道屏障功能、免疫调节、脂质代谢等关键通路,为CD的诊断、预后与治疗靶点开发提供潜在方向。
Biomarker定位
明确的Biomarker类型为组织特异性易感基因,筛选逻辑为:基于不同人群的GWAS数据,整合对应种族与组织的eQTL数据,通过TWAS预测基因表达与CD的关联,然后通过多个独立研究的重复筛选验证其可靠性;验证逻辑包括跨研究重复、跨组织重复、功能实验验证(部分基因)。
研究过程详述
Biomarker来源包括回肠、结肠、肠道免疫细胞、血液免疫细胞、全血的基因表达数据,其中回肠与结肠的基因表达数据来自肠道病变组织或正常组织,血液数据来自全血或免疫细胞亚群;验证方法主要为多个独立TWAS研究的重复筛选,如ATG16L1在3项研究的回肠与血液中被重复筛选到,ERAP2在4项研究的回肠、结肠、血液中被重复筛选到;特异性与敏感性方面,不同Biomarker在不同人群中的关联强度不同,如TNFSF15在东亚人群(日本、韩国、中国)中与CD的关联更强,且与肛周病变等并发症相关(文献未明确提供具体P值,具有统计学显著性),在欧洲人群中则表现为保护作用;ERAP2在多个组织与人群中均有显著关联,具有较好的特异性;部分基因的功能实验验证显示,ATG16L1突变可导致潘氏细胞功能异常,破坏肠道屏障,增加细菌感染风险;NOD2可识别细菌胞壁成分,调节免疫应答。
核心成果提炼
总结了10余个在多个组织与人群中重复验证的CD易感基因,其中ATG16L1通过调控自噬与潘氏细胞功能参与肠道屏障维持,风险突变与回肠型CD及抗TNF治疗应答相关(P<0.05,文献未明确样本量);ERAP2参与抗原提呈,影响HLA-I类分子的配体库,与多种炎症性疾病相关(P<0.05,多研究重复验证);IL23R参与Th17细胞分化,是CD治疗的重要靶点,已有多个靶向IL23的单抗进入临床III期试验;NOD2是CD风险最高的易感基因之一,参与细菌识别与免疫调节(P<0.001,大样本GWAS验证);TNFSF15在东亚人群中与CD并发症相关,其抑制剂已进入UC的IIa期临床试验;创新性在于首次系统揭示了CD TWAS研究中组织特异性的易感基因与功能通路,明确了回肠脂质代谢与结肠细胞外囊泡在发病中的潜在作用,为后续Biomarker的验证与临床应用提供了基础。