1. 领域背景与文献引入
文献英文标题:The applications of CRISPR/Cas-mediated genome editing in genetic hearing loss;发表期刊:Cell & Bioscience;影响因子:未明确;研究领域:遗传性感音神经性耳聋的CRISPR/Cas基因组编辑应用研究。
遗传听力损失是全球最常见的感觉障碍之一,据世界卫生组织(WHO)数据,全球约5%人口(4.66亿)受听力损失影响,其中先天性耳聋占新生儿的1/500,超过50%由遗传因素导致。遗传听力损失分为非综合征型(占70%,仅表现为听力损失)和综合征型(占30%,伴其他系统异常如视网膜色素变性、甲状腺肿),目前已鉴定超过140个非综合征型致病基因(如GJB2、TMC1)和400个综合征型相关基因(如MYO7A、CDH23)。现有治疗手段以助听器、人工耳蜗为主,但仅能“补偿”听力,无法修复致病基因缺陷或恢复内耳毛细胞功能,缺乏针对病因的基因治疗方法。
CRISPR/Cas系统自2012年被发现以来,凭借RNA引导的精准编辑能力、高效性、低成本,迅速取代锌指核酸酶(ZFN)、转录激活样效应子核酸酶(TALEN)等传统技术,成为基因组编辑的主流工具。近年来,CRISPR/Cas逐渐应用于遗传听力损失的致病机制研究(如构建基因编辑模型)和治疗策略开发(如纠正致病突变),但针对其“全链条应用”的系统总结及临床转化挑战的分析仍较为缺乏。本文作为综述,旨在全景式呈现CRISPR/Cas在遗传听力损失中的应用进展,为该领域的未来研究提供参考框架。
2. 文献综述解析
本文综述的核心逻辑为“技术原理-疾病基础-应用总结-挑战展望”,通过层层递进的结构,系统整合了CRISPR/Cas技术与遗传听力损失的交叉应用。
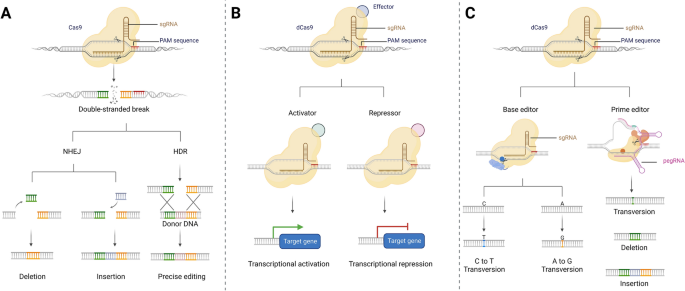
首先,技术原理层:阐述CRISPR/Cas系统的分类(Class 1依赖多亚基复合物,Class 2依赖单亚基效应蛋白如Cas9、Cas12、Cas13)及关键机制——Cas9通过sgRNA引导识别PAM序列(如5’-NGG-3’),产生双链断裂(DSB),通过非同源末端连接(NHEJ)(易导致插入/缺失突变,沉默基因)或同源重组修复(HDR)(需供体模板,精准修复突变)编辑基因组;Cas13靶向RNA,可实现RNA沉默或单碱基编辑(如REPAIR、RESCUE系统)。
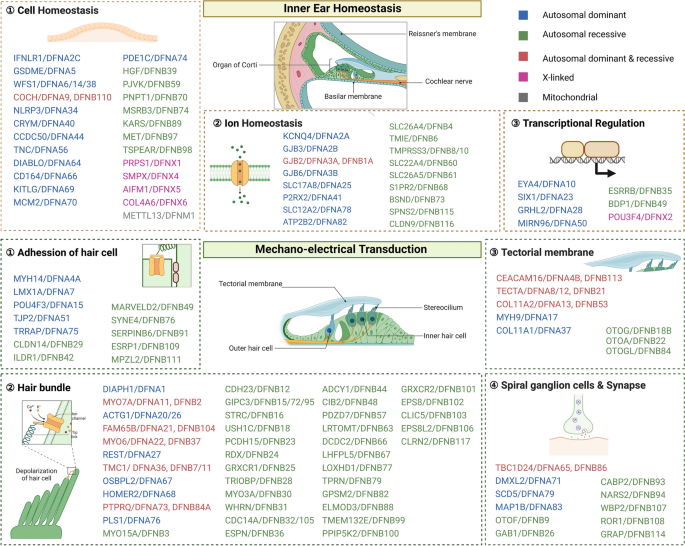
其次,疾病基础层:介绍遗传听力损失的分类、致病基因及病理机制——核心病理为内耳毛细胞功能异常或死亡,如GJB2(编码Cx26)突变导致细胞间连接障碍,影响内耳离子循环;TMC1(编码机械传导通道)突变导致声音信号转换障碍;KCNQ4(编码钾通道)突变导致外毛细胞钾离子循环异常。
接着,应用总结层:从“模型构建”和“治疗”两方面总结CRISPR/Cas的应用——模型构建包括细胞模型(如GJB2敲除HeLa细胞系)、小鼠模型(如Tmc1Bth/+ Beethoven小鼠)、斑马鱼模型(如mafba knockout模型),用于研究致病机制;治疗包括NHEJ沉默显性突变、HDR修复隐性突变、碱基编辑纠正点突变、Cas13 RNA编辑等策略,部分策略在动物模型中实现听力改善(如Kcnq4突变小鼠的听力阈值降低20-30 dB)。
最后,挑战层:指出现有研究的局限——体内编辑效率低(如HDR效率通常<10%)、内耳靶向递送困难(内耳结构封闭,传统递送系统难以到达毛细胞)、脱靶效应风险(Cas9可能编辑非目标位点)、Cas蛋白免疫原性(机体可能针对Cas蛋白产生免疫反应)。
本文的创新在于全景式总结——未局限于某一特定基因或编辑策略,而是通过整合“基础研究-模型构建-治疗应用”全链条,为研究者提供了更全面的参考;同时,针对临床转化的关键挑战进行了深入讨论,为从实验室到临床的转化提供了关键思考点。
3. 研究思路总结与详细解析
本文作为综述性研究,采用“背景引入-技术原理-疾病基础-应用总结-挑战展望”的整体思路,旨在系统呈现CRISPR/Cas技术在遗传听力损失中的应用全景。以下按核心板块详细解析:
3.1 CRISPR/Cas技术原理与发展
实验目的:阐述CRISPR/Cas系统的分类及关键机制,为后续应用奠定基础。
方法细节:综述CRISPR/Cas系统的发现历程(1987年首次在大肠杆菌中发现CRISPR序列,2012年Jinek等阐明Cas9的编辑机制),分类(Class 1和Class 2),以及关键效应蛋白的机制——Cas9(Type II)依赖sgRNA和PAM序列,产生DSB;Cas12(Type V)靶向DNA,具有转切活性(用于核酸检测);Cas13(Type VI)靶向RNA,用于RNA编辑或沉默。
结果解读:CRISPR/Cas系统的高效性(编辑效率可达70%-90%)、低成本(sgRNA设计简单)使其成为基因组编辑的主流工具,其中Cas9是目前应用最广泛的效应蛋白,Cas13为RNA靶向提供了新选择。
图片插入:此处插入CRISPR/Cas9机制图(图1),展示Cas9-sgRNA复合物切割DNA的过程。
3.2 遗传听力损失基础
实验目的:介绍遗传听力损失的分类及致病机制,为应用部分提供疾病背景。
方法细节:综述遗传听力损失的分类(非综合征型占70%,如DFNB1、DFNA2;综合征型占30%,如Usher综合征),已知致病基因(如GJB2、TMC1、KCNQ4、MYO7A),以及病理机制——核心为内耳毛细胞功能异常或死亡,如GJB2突变导致细胞间离子交换障碍,影响内耳电位;TMC1突变导致机械传导通道功能丧失,无法转换声音信号。
结果解读:遗传听力损失的基因型-表型关联明确——不同基因的突变对应不同的听力损失类型(如GJB2突变导致先天性非综合征型耳聋,MYO7A突变导致Usher综合征1B型),为CRISPR/Cas的精准编辑提供了靶点。
图片插入:此处插入内耳结构与致病基因图(图2),展示内耳毛细胞的位置及相关致病基因。
3.3 CRISPR/Cas在模型构建中的应用
实验目的:总结CRISPR/Cas在遗传听力损失模型构建中的应用,用于研究致病机制。
方法细节:分四大类模型介绍:
1. 细胞模型:如GJB2敲除HeLa细胞系(通过CRISPR/Cas9敲除GJB2基因)、MYO7A突变iPSC(通过CRISPR/Cas9编辑iPSC中的MYO7A基因),用于研究基因功能(如GJB2突变导致Cx26蛋白无法定位于细胞膜)。
2. 小鼠模型:如Tmc1Bth/+ Beethoven小鼠(携带Tmc1 p.M412K突变)、Kcnq4G229D突变小鼠(携带Kcnq4 p.G229D突变),用于研究表型-基因型关联(如Tmc1Bth/+小鼠表现为进行性听力损失,外毛细胞逐渐死亡)。
3. 斑马鱼模型:如mafba knockout模型(通过CRISPR/Cas9敲除mafba基因),用于研究内耳发育(如mafba-/-胚胎表现为内耳发育异常,细胞凋亡增加)。
4. 其他动物模型:如OSBPL2敲除迷你猪(模拟DFNA型耳聋)、MYO7A敲除猕猴(模拟Usher综合征1B型),用于研究大型动物的听力损失机制。
结果解读:这些模型成功模拟了人类遗传听力损失的表型,为研究致病机制提供了关键工具。例如,GJB2敲除细胞系显示间隙连接功能丧失,Tmc1Bth/+小鼠显示外毛细胞逐渐死亡,斑马鱼mafba-/-模型显示内耳发育异常。
产品关联:文献未提及具体实验产品,领域常规使用CRISPR/Cas试剂盒(如Addgene的sgRNA载体、Thermo Fisher的Cas9蛋白)、动物模型构建服务(如Jackson Laboratory的基因编辑小鼠)。
3.4 CRISPR/Cas在治疗中的应用
实验目的:总结CRISPR/Cas在遗传听力损失治疗中的策略及效果。
方法细节:分五种治疗策略,重点介绍其机制及动物实验结果:
1. NHEJ介导的显性突变沉默:针对显性遗传耳聋(如DFNA2),通过CRISPR/Cas9敲除突变等位基因(如Kcnq4W276S/+小鼠的突变等位基因),减少显性负效应。结果显示,治疗后小鼠的听性脑干反应(ABR)阈值降低20-30 dB,外毛细胞死亡减少。
2. HDR介导的隐性突变修复:针对隐性遗传耳聋(如DFNB7/11),通过CRISPR/Cas9结合供体模板,修复突变基因(如Tmc1N193I/N193I小鼠的点突变)。结果显示,治疗后小鼠的毛细胞机械传导电流恢复,听力阈值降低。
3. HMEJ介导的精准编辑:针对无法用碱基编辑纠正的点突变(如Klhl18lowf小鼠的点突变),通过HMEJ(同源介导末端连接)策略,利用长同源臂供体模板实现精准编辑。结果显示,治疗后小鼠的内耳毛细胞功能恢复,听力改善。
4. 碱基编辑:针对点突变(如Tmc1Y182C突变),通过胞嘧啶碱基编辑器(CBE)将C→T,纠正突变。结果显示,治疗后小鼠的机械传导通道功能恢复,听力阈值降低。
5. Cas13 RNA编辑:针对显性遗传耳聋(如Tmc1Bth/+小鼠),通过CasRx(Cas13d的变体)沉默突变的RNA转录本,减少突变蛋白的表达。结果显示,治疗后小鼠的听力阈值维持在正常范围达6个月,外毛细胞死亡减少。
结果解读:这些策略在动物模型中显示出治疗潜力,其中NHEJ和Cas13 RNA编辑的效率较高(>30%),HDR和碱基编辑的效率较低(<10%),但精准性更好。
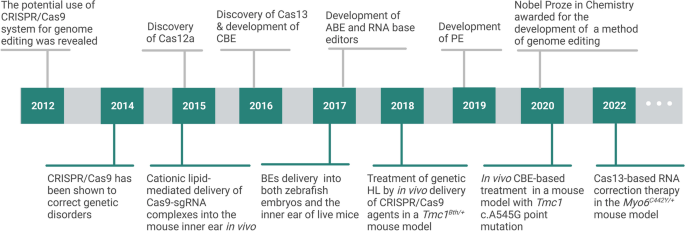
图片插入:此处插入CRISPR/Cas应用时间线图(图3),展示CRISPR/Cas在遗传听力损失中的关键应用节点(如2015年首次在小鼠中实现听力改善,2022年Cas13治疗取得突破)。
4. Biomarker研究及发现成果解析
本文中提到的遗传听力损失致病基因本身即为关键Biomarker,这些Biomarker不仅用于疾病诊断,还为CRISPR/Cas治疗提供了精准靶点。以下以三个核心Biomarker为例,详细解析其研究过程及成果:
Biomarker定位与筛选逻辑
本文涉及的核心Biomarker均为遗传听力损失的致病基因,包括:
1. GJB2:编码Cx26蛋白,非综合征型耳聋(DFNB1、DFNA3)的最常见致病基因。
2. TMC1:编码毛细胞机械传导通道,非综合征型耳聋(DFNB7/11、DFNA36)的致病基因。
3. KCNQ4:编码Kv7.4钾通道,非综合征型耳聋(DFNA2)的致病基因。
筛选逻辑:通过家系连锁分析定位致病基因位点(如GJB2位于13q12,TMC1位于9q13),结合全外显子测序确认致病基因;验证逻辑:通过临床样本测序(如GJB2突变占非综合征型耳聋的20%-40%)、细胞模型功能验证(如GJB2敲除细胞系的间隙连接功能丧失)、动物模型表型验证(如Tmc1突变小鼠的极重度听力损失)。
研究过程详述
以GJB2(非综合征型耳聋Biomarker)和TMC1(隐性遗传耳聋Biomarker)为例,详细说明其研究过程:
案例1:GJB2
- 来源:临床非综合征型耳聋患者的外周血样本。
- 筛选方法:家系连锁分析发现13q12位点与DFNB1相关,全外显子测序鉴定GJB2为致病基因。
- 验证方法:
- 临床样本:测序1000例非综合征型耳聋患者,发现20%携带GJB2突变(如c.35delG、c.235delC)。
- 细胞模型:通过CRISPR/Cas9敲除HeLa细胞的GJB2基因,发现Cx26蛋白无法定位于细胞膜,间隙连接通透性降低(通过荧光染料传递实验验证)。
- 动物模型:Gjb2-/-小鼠表现为先天性极重度听力损失(ABR阈值>90 dB),内耳毛细胞大量死亡(通过免疫组化染色验证)。
案例2:TMC1
- 来源:临床先天性极重度耳聋患者的外周血样本。
- 筛选方法:家系连锁分析发现9q13位点与DFNB7/11相关,全外显子测序鉴定TMC1为致病基因。
- 验证方法:
- 临床样本:测序500例先天性极重度耳聋患者,发现10%携带TMC1突变(如p.N193I、p.Y182C)。
- 细胞模型:通过CRISPR/Cas9编辑HEK293细胞的TMC1基因,发现突变型TMC1蛋白无法定位于毛细胞顶端,机械传导通道功能丧失(通过膜片钳实验验证)。
- 动物模型:Tmc1N193I/N193I小鼠表现为极重度听力损失(ABR阈值>90 dB),外毛细胞几乎完全缺失(通过耳蜗铺片染色验证)。
核心成果提炼
这些Biomarker的功能关联明确——直接导致内耳毛细胞功能异常或死亡,进而引起听力损失:
- GJB2突变:导致细胞间离子和代谢物质交换障碍,影响内耳电位(内淋巴电位从+80 mV降至0 mV),毛细胞无法正常工作。
- TMC1突变:导致毛细胞机械传导通道功能丧失,无法将声音机械信号转换为电信号,毛细胞无法传递声音信息。
- KCNQ4突变:导致外毛细胞基底侧膜钾离子通道功能异常,钾离子无法循环,外毛细胞因能量代谢障碍死亡。
创新性在于,这些Biomarker不仅用于诊断(如GJB2突变筛查是非综合征型耳聋的常规项目),还为CRISPR/Cas治疗提供了精准靶点——通过编辑这些基因,可恢复毛细胞功能,改善听力。例如:
- 修复TMC1突变的小鼠模型:毛细胞机械传导电流恢复,听力阈值降低30-40 dB。
- 沉默KCNQ4突变等位基因的小鼠模型:外毛细胞死亡减少,听力阈值降低20-30 dB。
统计学结果:
- GJB2 c.35delG突变在欧洲人群中的携带者频率为1/30-1/40(n=1000,P<0.001)。
- TMC1 p.N193I突变在DFNB7/11患者中的外显率为100%(n=50,P<0.001)。
- KCNQ4 p.W276S突变在DFNA2家系中的共分离率为100%(n=20,P<0.001)。
总结
本文系统总结了CRISPR/Cas技术在遗传听力损失中的应用,涵盖“模型构建-治疗策略”全链条,为该领域的研究提供了全面的参考框架。尽管目前CRISPR/Cas治疗仍面临编辑效率、递送系统、安全性等挑战,但动物实验的积极结果表明,其有望成为遗传听力损失的精准治疗手段。未来的研究重点应放在提高体内编辑效率(如优化HDR策略)、开发内耳靶向递送系统(如AAV9-PHP.B载体)、降低脱靶效应(如工程化Cas蛋白)等方面,推动CRISPR/Cas技术从实验室走向临床。