1. 领域背景与文献引入
文献英文标题:Ubiquitinated AIF is a major mediator of hypoxia-induced mitochondrial dysfunction and pulmonary artery smooth muscle cell proliferation;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:低氧性肺动脉高压的分子机制研究
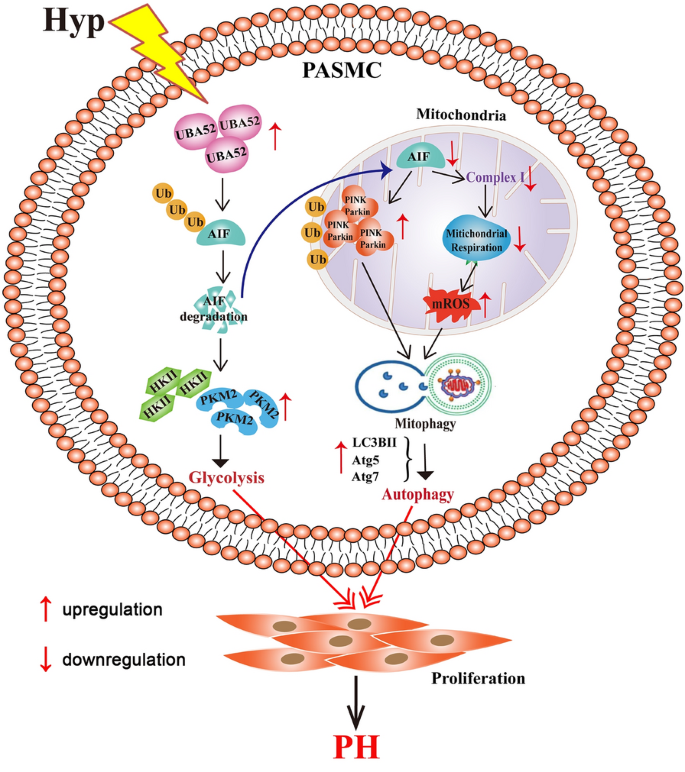
肺动脉高压(PH)是一类致死性心血管呼吸系统疾病,以肺动脉平均压力进行性升高、劳力性呼吸困难为特征,最终导致右心衰竭与死亡。低氧是诱导PH肺血管病理改变的常见诱因,通过调控细胞增殖、焦亡、自噬等多种细胞过程诱导肺血管重构,其中肺动脉平滑肌细胞(PASMCs)过度增殖是肺血管重构的核心驱动因素,但调控该病理过程的具体分子细胞机制仍不明确。线粒体作为细胞能量代谢的核心细胞器,其稳态维持对细胞功能至关重要,线粒体呼吸链损伤会导致氧化磷酸化(OXPHOS)障碍、活性氧(ROS)生成增加,参与多种疾病的发生发展。线粒体自噬通过选择性清除受损线粒体维持细胞稳态,但其在低氧性PH中的病理生理机制尚未阐明。凋亡诱导因子(AIF)是定位于线粒体膜间隙的氧化还原酶,参与维持线粒体结构与呼吸链复合物功能,在肿瘤、代谢性疾病等多种疾病中发挥作用,但在低氧诱导的PH线粒体功能调控中的作用尚未见报道,这一研究空白为本研究提供了学术必要性,即明确AIF在低氧性PH中的作用及调控机制,为PH的治疗提供新靶点。
2. 文献综述解析
作者从“PH的核心病理机制、线粒体功能与自噬的作用、AIF的生物学功能”三个维度对现有研究进行分类评述,系统梳理了领域内的研究进展与未解决问题。
现有研究显示,PH的核心病理特征是肺血管重构,PASMCs过度增殖是关键环节,低氧通过多通路调控这一过程,但具体分子靶点不明;线粒体呼吸链损伤会导致能量代谢异常与ROS生成增加,参与PH的发生,但低氧下线粒体功能的调控机制尚未明确;自噬在PH中的作用已有报道,但低氧诱导自噬的上游调控分子仍不清楚;AIF作为线粒体氧化还原酶,在维持线粒体复合物I功能、OXPHOS中起重要作用,参与多种疾病,但在PH中的研究完全空白。现有研究的局限性在于,未将AIF与低氧下线粒体功能、自噬、PASMCs增殖关联起来,缺乏对AIF在PH中调控机制的研究。本研究的创新价值在于,首次揭示了低氧下UBA52介导的AIF泛素化下调机制,明确了AIF通过调控线粒体功能、自噬抑制PASMCs增殖的作用,填补了低氧性PH分子机制研究的空白,为PH的治疗提供了新的潜在靶点。
3. 研究思路总结与详细解析
本研究的核心目标是明确AIF在低氧诱导PH中的作用及调控机制,核心科学问题是低氧下AIF如何通过线粒体功能、自噬通路调控PASMCs增殖,技术路线遵循“模型构建→分子表达与定位分析→功能验证(细胞+动物)→调控机制解析→通路验证”的闭环逻辑,从细胞、动物、分子三个层面系统验证了AIF的功能与调控机制。
3.1 低氧模型构建与线粒体功能损伤验证
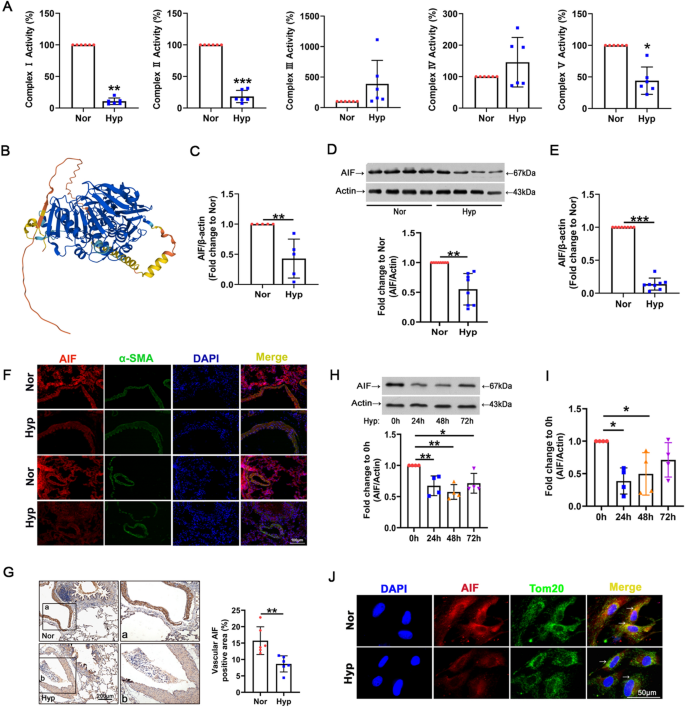
实验目的是验证低氧对线粒体呼吸链的损伤及AIF表达与定位的变化。方法细节上,构建小鼠低氧PH模型(吸入氧浓度FiO₂=0.12,持续28天),分离培养大鼠PASMCs并置于3% O₂的低氧培养箱中处理;采用线粒体呼吸链复合物活性检测试剂盒检测肺组织中线粒体复合物I-V的活性,通过qRT-PCR、蛋白质免疫印迹(Western blotting)检测AIF的mRNA与蛋白表达水平,利用免疫荧光、免疫组化技术检测AIF在肺组织与PASMCs中的定位。结果解读显示,低氧显著降低线粒体复合物I、II、V的活性,其中复合物I的活性下降最为显著(n=6,P<0.001);低氧处理后,肺组织与PASMCs中AIF的mRNA与蛋白表达均显著下调(n=8,P<0.01),AIF主要定位于PASMCs的线粒体中,低氧后线粒体与细胞质中的AIF表达均明显降低。实验所用关键产品:Abcam的AIF抗体(货号ab32516)、Solarbio的线粒体呼吸链复合物I-V活性检测试剂盒。
3.2 AIF对线粒体能量代谢与氧化应激的调控
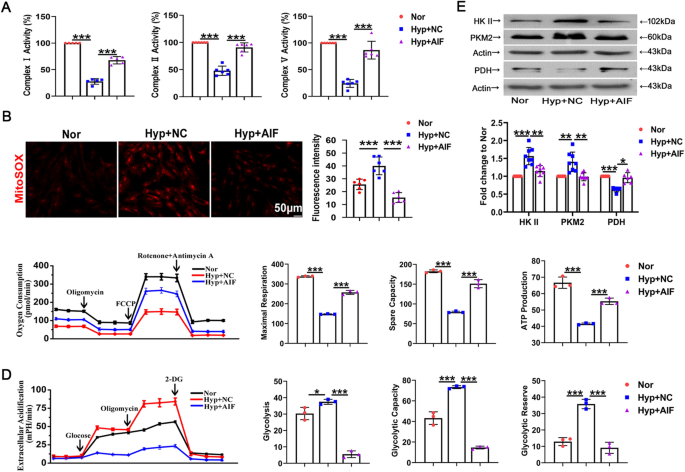
实验目的是验证AIF对低氧诱导的线粒体能量表型转换与氧化应激的调控作用。方法细节上,构建AIF过表达质粒并转染PASMCs,采用Seahorse细胞外通量分析仪检测细胞的氧消耗率(OCR,反映线粒体氧化磷酸化水平)与细胞外酸化率(ECAR,反映糖酵解水平),利用MitoSOX Red荧光探针检测线粒体ROS生成水平,通过蛋白质免疫印迹检测糖酵解关键酶(HK II、PKM2)与氧化磷酸化关键酶(PDH)的表达。结果解读显示,AIF过表达可显著恢复低氧下线粒体复合物I、II、V的活性(n=5-6,P<0.05);低氧诱导的线粒体ROS生成增加被AIF过表达有效抑制(n=6,P<0.01);低氧导致的最大呼吸能力、线粒体ATP生成能力下降在AIF过表达后明显恢复(n=3,P<0.05),低氧激活的糖酵解通路被AIF过表达阻断(n=3,P<0.01);低氧诱导的HK II、PKM2表达上调及PDH表达下调,均被AIF过表达逆转(n=6-8,P<0.05)。实验所用关键产品:Seahorse细胞外通量分析仪、Invitrogen的MitoSOX Red(货号M36008)、Cell Signaling Technology的HK II抗体(货号2867)。
3.3 AIF对低氧诱导的线粒体自噬与自噬的调控
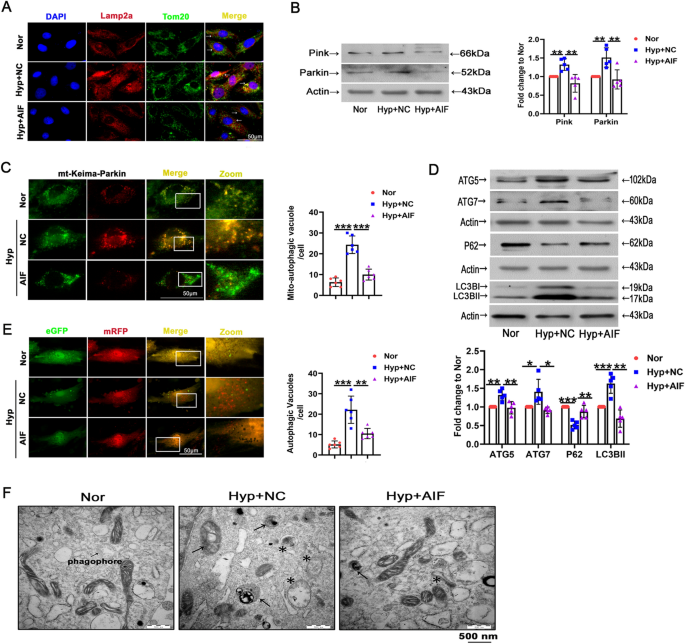
实验目的是明确AIF在低氧诱导的线粒体自噬与整体自噬中的调控作用。方法细节上,采用免疫荧光技术检测线粒体标记Tom20与溶酶体标记Lamp2a的共定位情况,通过蛋白质免疫印迹检测线粒体自噬标记(Pink、Parkin)与自噬核心标记(LC3BII、P62、ATG5/7)的表达,转染pMitophagy Keima-Red mPark2质粒监测线粒体自噬通量,利用透射电镜(TEM)观察PASMCs中自噬体与自噬溶酶体的数量变化。结果解读显示,低氧诱导Tom20与Lamp2a的共定位显著增加(线粒体自噬增强),AIF过表达可明显减少该共定位(n=4,P<0.05);低氧诱导的Pink、Parkin表达上调被AIF过表达有效抑制(n=5,P<0.01);低氧处理后Keima红色荧光比例增加(线粒体向溶酶体转运增加),AIF过表达可阻断这一效应(n=5,P<0.01);低氧诱导的LC3BII表达上调、P62表达下调、ATG5/7表达上调,均被AIF过表达逆转(n=5,P<0.05);透射电镜显示低氧下PASMCs中自噬体与自噬溶酶体数量显著增加,AIF过表达可减少其数量(n=3)。实验所用关键产品:Abcam的Tom20抗体(货号ab56783)、Lamp2a抗体(货号ab18528)、MBL的pMitophagy Keima-Red mPark2质粒(货号AM-V0259M)。
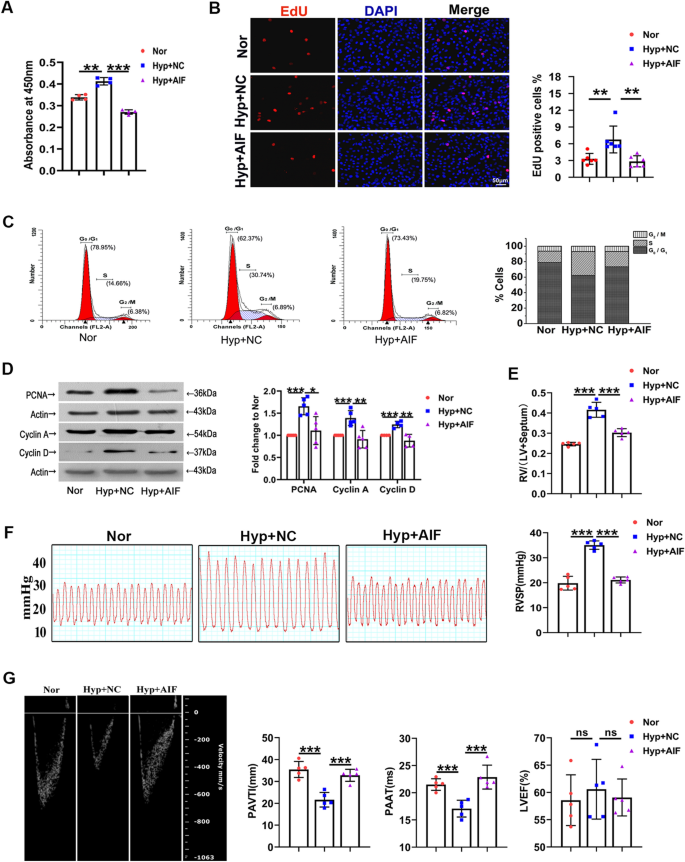
3.4 AIF对PASMCs增殖与体内肺血管重构的调控
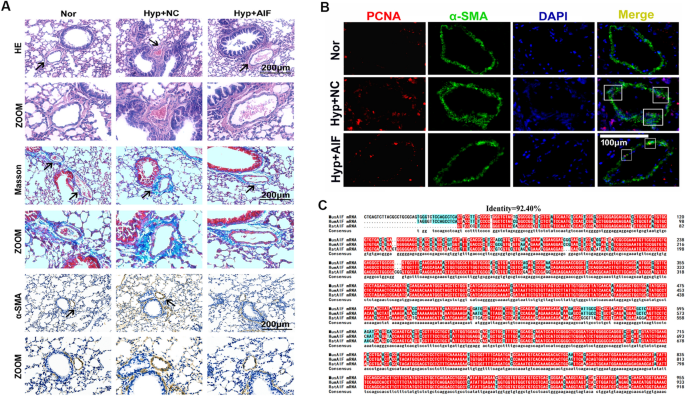
实验目的是验证AIF对低氧诱导的PASMCs增殖及体内肺血管重构的抑制作用。方法细节上,采用CCK8实验、EdU掺入实验检测PASMCs的增殖活力,通过流式细胞术检测细胞周期分布,利用蛋白质免疫印迹检测增殖标记物(PCNA、Cyclin A、Cyclin D)的表达;构建AAV5-AIF过表达载体,经滴鼻感染小鼠后置于低氧环境中处理28天,采用超声心动图、右心室收缩压(RVSP)检测评估PH的病理指标,通过H&E染色、Masson染色、免疫组化染色观察肺血管重构情况。结果解读显示,低氧诱导的PASMCs活力增加被AIF过表达显著抑制(n=4,P<0.05);低氧诱导的EdU阳性细胞比例增加被AIF过表达有效阻断(n=6,P<0.01);低氧诱导的G₂/M+S期细胞比例增加被AIF过表达抑制(n=3,P<0.05);低氧诱导的PCNA、Cyclin A、Cyclin D表达上调被AIF过表达逆转(n=4-5,P<0.05);体内实验中,AIF过表达可显著抑制低氧诱导的右心室肥厚(RV/LV+S比值降低,n=6,P<0.01)、RVSP升高(n=5,P<0.01),改善超声心动图中的肺动脉血流指标(PAVTI、PAAT恢复,n=6,P<0.05);低氧诱导的肺血管中膜增厚、PCNA阳性细胞增加被AIF过表达有效抑制(n=5,P<0.01)。实验所用关键产品:RiboBio的EdU试剂、GeneChem的AAV5载体、BD FACSCalibur流式细胞仪。
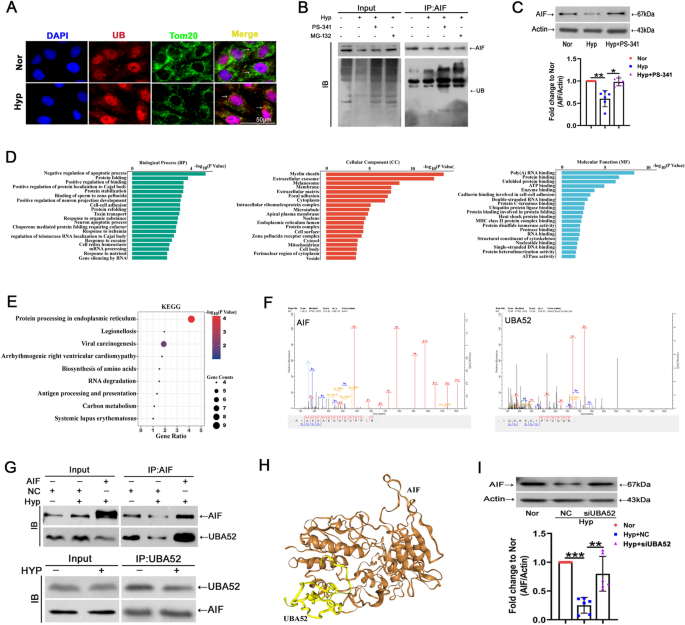
3.5 AIF泛素化调控机制解析
实验目的是明确低氧下AIF表达下调的泛素化调控机制。方法细节上,采用免疫荧光技术检测泛素(UB)与线粒体标记Tom20的共定位情况,通过免疫共沉淀(Co-IP)结合蛋白质免疫印迹检测AIF的泛素化水平,利用蛋白酶体抑制剂PS-341处理验证泛素化介导的AIF降解,通过质谱分析筛选AIF的相互作用蛋白,采用Co-IP验证AIF与UBA52的相互作用,利用siRNA沉默UBA52后检测AIF的表达变化。结果解读显示,低氧下PASMCs中UB与Tom20的共定位显著增加(线粒体泛素化增强,n=3);Co-IP实验检测到AIF的泛素化条带,PS-341处理可显著恢复低氧下AIF的蛋白表达水平(n=6,P<0.01);质谱分析鉴定到AIF的相互作用蛋白UBA52,Co-IP实验进一步验证了两者的直接相互作用;分子对接结果显示AIF与UBA52存在明确的结合位点;siRNA沉默UBA52后,低氧下PASMCs中AIF的蛋白表达水平显著升高(n=6,P<0.01)。实验所用关键产品:Abcam的UBA52抗体(货号ab109227)、Selleck的PS-341(货号S1013)、GenePharma的siRNA。
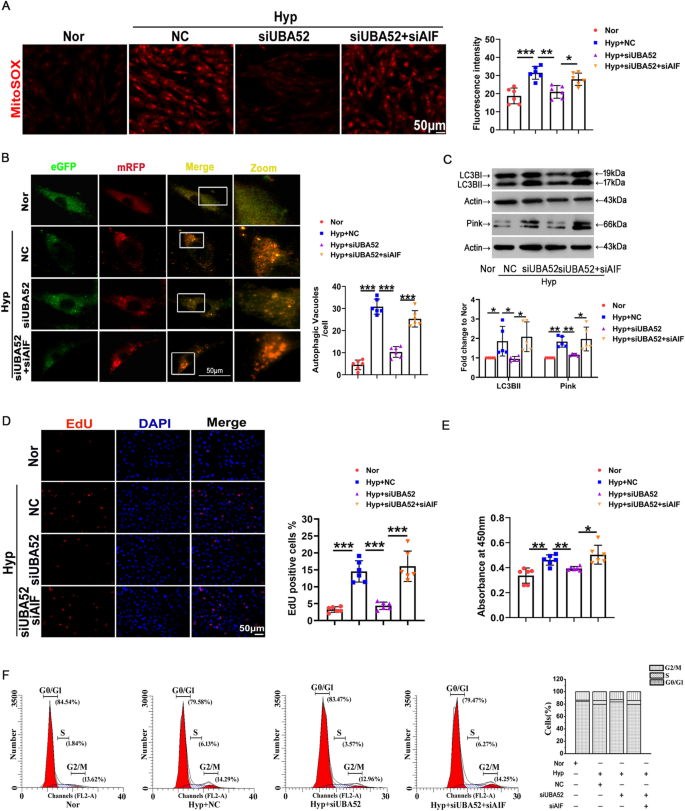
3.6 UBA52/AIF通路功能验证
实验目的是验证UBA52/AIF通路在低氧诱导的线粒体功能障碍、自噬激活与PASMCs增殖中的核心作用。方法细节上,共转染siUBA52与siAIF至PASMCs,采用MitoSOX Red检测线粒体ROS生成水平,转染eGFP-mRFP-LC3质粒检测自噬通量,通过蛋白质免疫印迹检测LC3BII、Pink的表达,利用CCK8实验、EdU掺入实验、流式细胞术检测细胞增殖与周期分布。结果解读显示,siUBA52可显著抑制低氧诱导的线粒体ROS生成增加,而共转染siAIF可逆转该效应(n=6,P<0.01);siUBA52可减少低氧诱导的自噬体形成,共转染siAIF可逆转该效应(n=6,P<0.01);siUBA52可抑制低氧诱导的LC3BII、Pink表达上调,共转染siAIF可逆转该效应(n=5,P<0.05);siUBA52可显著抑制低氧诱导的PASMCs活力增加、EdU阳性细胞比例增加、G₂/M+S期细胞比例增加,共转染siAIF可逆转该效应(n=6,P<0.01)。实验所用关键产品:Beyotime的eGFP-mRFP-LC3腺病毒(货号C3011)。
4. Biomarker研究及发现成果
本研究中鉴定的核心Biomarker为凋亡诱导因子(AIF),属于线粒体氧化还原酶类分子,其筛选与验证遵循“低氧模型中AIF表达异常→细胞功能验证AIF对线粒体、自噬、增殖的调控→体内验证AIF对肺血管重构的抑制→泛素化机制解析UBA52对AIF的调控”的完整逻辑链条,为低氧性PH的治疗提供了新的潜在靶点。
AIF的来源为肺动脉平滑肌细胞(PASMCs)的线粒体膜间隙与细胞质,验证方法包括qRT-PCR、蛋白质免疫印迹检测其在低氧下的表达变化,免疫荧光、免疫组化检测其细胞定位,细胞功能实验验证其对线粒体代谢、自噬、细胞增殖的调控作用,体内动物模型验证其对低氧性PH肺血管重构的抑制效应。特异性方面,AIF的下调与低氧诱导的线粒体复合物I损伤、自噬激活、PASMCs过度增殖直接相关,是低氧性PH病理过程中的关键调控分子;敏感性方面,低氧处理后AIF的mRNA与蛋白表达均显著下调(n=8,P<0.01),可灵敏反映低氧对线粒体功能的损伤。核心成果方面,AIF可作为低氧性PH的潜在治疗靶点,其泛素化下调介导了低氧诱导的线粒体功能障碍、自噬激活与PASMCs增殖,本研究首次揭示了UBA52介导的AIF泛素化调控机制,为PH的治疗提供了新的分子靶点与干预策略,同时为线粒体功能调控在PH中的作用提供了新的理论依据。