1. 领域背景与文献引入
文献英文标题:Targeting CD47/SIRPα as a therapeutic strategy, where we are and where we are headed;发表期刊:Biomarker Research;影响因子:未公开;研究领域:肿瘤免疫治疗(先天免疫检查点CD47/SIRPα轴靶向治疗)
肿瘤免疫治疗自2011年CTLA4抑制剂获批以来,历经PD-1/PD-L1抑制剂的临床突破,已成为多种恶性肿瘤的标准治疗方案,但仅30%左右的实体瘤患者对单药免疫检查点抑制剂产生持久响应,部分患者甚至出现免疫治疗相关超进展,其机制尚未完全明确。领域共识:巨噬细胞作为先天免疫系统中最丰富的细胞亚群,通过吞噬作用直接清除肿瘤细胞,同时启动适应性免疫应答,但其功能常被肿瘤细胞的“免疫逃逸信号”抑制。CD47作为关键的“别吃我”信号分子,通过与巨噬细胞表面的SIRPα结合,激活胞内磷酸酶信号通路,阻断吞噬作用,目前已成为肿瘤免疫治疗的新兴靶点。截至2022年,多款CD47/SIRPα靶向药物进入临床研究,但仍存在血液毒性显著、实体瘤疗效有限、联合治疗策略不明确等核心问题,亟需全面梳理现有研究进展,为后续药物开发提供方向。本文献针对上述研究空白,系统综述了CD47/SIRPα轴的生物学机制及各类靶向药物的研发现状,为该领域的临床转化提供全景式参考。
2. 文献综述解析
本文献为系统综述,作者按靶向CD47/SIRPα轴的药物类型(单克隆抗体、融合蛋白、双特异性抗体、细胞治疗、小分子/miRNA)分类,全面总结了该领域的临床前与临床研究进展,重点分析了各类药物的作用机制、疗效数据及安全性挑战,为该领域的后续研究提供了结构化参考框架。
现有研究可分为六大类:抗CD47单克隆抗体是最早进入临床的药物类型,其中Magrolimab作为首款进入III期的药物,联合利妥昔单抗在复发/难治性非霍奇金淋巴瘤中实现50%的客观缓解率(ORR),联合阿扎胞苷在急性髓系白血病(AML)中客观缓解率达65%,但存在剂量依赖性贫血等血液毒性;SIRPα-Fc融合蛋白通过突变优化提升与CD47的亲和力,Evorpacept联合利妥昔单抗在非霍奇金淋巴瘤中客观缓解率达63.6%,但单独使用无抗肿瘤活性;抗SIRPα单抗需联合调理素抗体才能有效诱导吞噬作用,目前处于早期临床阶段;双特异性抗体通过同时靶向CD47与肿瘤抗原或免疫检查点,实现肿瘤选择性靶向,降低对正常细胞的毒性,如NI1701(CD47/CD19双抗)在临床前研究中显示出比单药联合更强的肿瘤杀伤活性;CAR-T/M细胞治疗处于临床前阶段,初步显示出实体瘤治疗潜力;小分子与miRNA通过下调CD47或SIRPα表达发挥作用,如RRx-001可同时下调肿瘤细胞CD47与巨噬细胞SIRPα,目前处于III期临床。现有研究的局限性主要包括:抗CD47单抗的血液毒性限制剂量提升,SIRPα-Fc依赖联合治疗才能发挥活性,双特异性抗体与细胞治疗的安全性与疗效仍需临床验证,实体瘤中CD47靶向治疗的响应率较低。
通过对比现有研究的未解决问题,本文献的创新价值凸显:与既往综述仅聚焦单克隆抗体或融合蛋白不同,本文首次全面覆盖了CD47/SIRPα轴的所有靶向策略,包括新兴的CAR-M、小分子与miRNA疗法,同时整合了截至2022年的最新临床数据,详细分析了各类药物的安全性与疗效差异,为不同药物的开发方向提供了针对性参考,填补了该领域全景式综述的空白。
3. 研究思路总结与详细解析
本文献的研究目标是系统梳理CD47/SIRPα轴的生物学机制及各类靶向药物的研发现状,核心科学问题是如何通过靶向CD47/SIRPα轴实现安全有效的肿瘤免疫治疗,技术路线为“生物学机制阐述→各类靶向药物分类综述→临床数据与安全性分析→未来方向展望”的逻辑闭环。
3.1 CD47/SIRPα轴生物学机制概述
实验目的:明确CD47与SIRPα的分子结构及相互作用机制,为靶向策略提供理论基础。
方法细节:通过文献调研,整合CD47与SIRPα的蛋白结构解析、表达分布及信号通路研究数据。
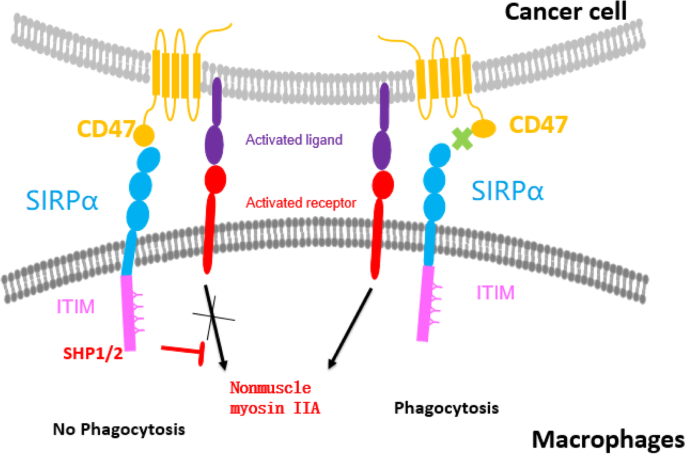
结果解读:CD47是跨膜糖蛋白,表达于多种正常细胞与肿瘤细胞表面;SIRPα主要表达于髓系细胞表面,其胞内段含免疫受体酪氨酸抑制基序(ITIM)。CD47的IgV结构域与SIRPα的D1结构域结合后,激活SHP-1/2磷酸酶,阻断巨噬细胞的吞噬信号通路。肿瘤细胞通过高表达CD47逃避巨噬细胞吞噬,而钙网蛋白(CRT)作为“吃我”信号,与CD47的平衡决定了肿瘤细胞是否被吞噬。
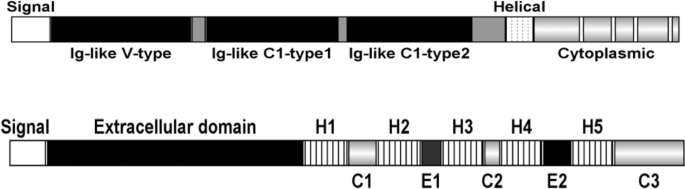
产品关联:文献未提及具体实验产品,领域常规使用免疫组化(IHC)抗体、流式细胞术抗体检测CD47与SIRPα表达。
3.2 抗CD47单克隆抗体研发现状解析
实验目的:总结抗CD47单抗的作用机制、临床前优化及临床研究数据。
方法细节:整合10余款进入临床的抗CD47单抗的临床研究数据,包括剂量爬坡、联合治疗、安全性终点等核心信息。
结果解读:抗CD47单抗通过阻断CD47/SIRPα相互作用,同时结合Fc段激活FcγR信号,增强巨噬细胞吞噬作用,部分单抗可直接诱导肿瘤细胞凋亡。Magrolimab作为首款进入III期的药物,在复发/难治性AML中联合阿扎胞苷的客观缓解率为65%(n=34,文献未明确提供P值),但因血液毒性被FDA暂停部分研究;Ligufalimab(AK117)在I期临床中未出现剂量限制性毒性,无需预激剂量,在45mg/kg剂量下未诱导贫血;Lemzoparlimab(TJC4)因结合表位不同,对红细胞毒性较低,I期临床中30%患者出现轻度贫血。
产品关联:实验所用关键产品:Magrolimab(Hu5F9-G4)、Ligufalimab(AK117)、Lemzoparlimab(TJC4)等。
3.3 SIRPα-Fc融合蛋白研究进展
实验目的:分析SIRPα-Fc融合蛋白的亲和力优化、作用机制及临床疗效。
方法细节:总结6款进入临床的SIRPα-Fc融合蛋白的临床前优化数据与临床研究结果,重点关注联合治疗的疗效。
结果解读:野生型SIRPα与CD47的亲和力较弱,通过突变优化后的SIRPα-Fc(如Evorpacept)亲和力提升50000倍,联合利妥昔单抗在非霍奇金淋巴瘤中客观缓解率达63.6%(n=22,文献未明确提供P值),但单独使用无抗肿瘤活性;TTI-621/TTI-622基于野生型SIRPα构建,TTI-622在复发/难治性淋巴瘤中客观缓解率为33%(n=30,文献未明确提供P值),但存在一过性贫血;IMM01因不结合红细胞,避免了“抗原 sink”效应,联合利妥昔单抗显示出协同抗肿瘤活性。
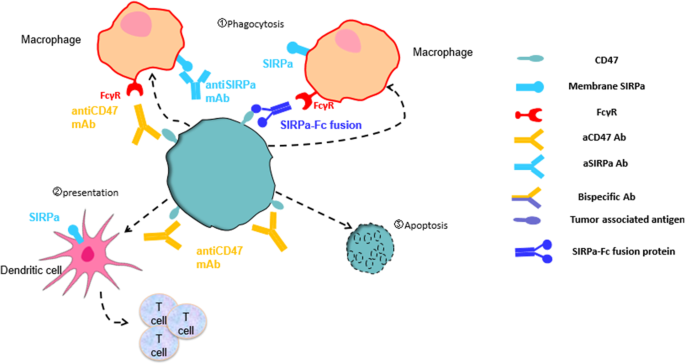
产品关联:实验所用关键产品:Evorpacept(ALX148)、TTI-621、TTI-622、IMM01等。
3.4 抗SIRPα单克隆抗体研究解析
实验目的:探讨抗SIRPα单抗的作用机制、特异性优化及临床潜力。
方法细节:总结进入临床的抗SIRPα单抗的设计策略,包括Fc段修饰、等位基因覆盖等,分析其临床前与早期临床数据。
结果解读:抗SIRPα单抗单独使用诱导吞噬作用较弱,需联合调理素抗体(如利妥昔单抗、西妥昔单抗)才能有效激活巨噬细胞吞噬;为避免髓系细胞毒性,抗SIRPα单抗多采用惰性Fc段;同时需针对SIRPα的主要等位基因(V1、V2、V8)设计,以覆盖大部分人群;目前OSE-172、CC-95251等处于早期临床阶段,安全性与疗效仍在评估中。
产品关联:文献未提及具体实验产品,领域常规使用重组SIRPα蛋白、髓系细胞系进行体外吞噬实验。
3.5 双特异性抗体及多靶点策略
实验目的:总结双特异性抗体的设计策略、肿瘤选择性及临床前数据。
方法细节:分类总结靶向CD47+肿瘤抗原、CD47+免疫检查点、SIRPα+其他靶点的双特异性抗体的临床前研究结果,分析其优势与挑战。
结果解读:双特异性抗体通过同时结合CD47与肿瘤抗原(如CD19、MSLN),实现肿瘤细胞选择性靶向,降低对正常细胞的毒性,如NI1701(CD47/CD19双抗)在临床前研究中对CD47+CD19+肿瘤细胞的杀伤活性显著高于单药联合;靶向CD47+PD-1/PD-L1的双特异性抗体(如HX009)可同时激活先天与适应性免疫,在I期临床中显示出跨瘤种的客观缓解;双特异性抗体目前处于早期临床阶段,其生产工艺与稳定性仍需优化。
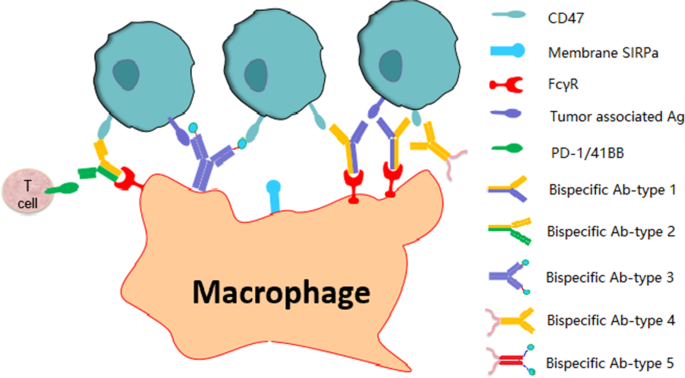
产品关联:实验所用关键产品:NI1701、HX009、IMM0306等。
3.6 CAR-T/M及其他新兴策略(小分子、miRNA)
实验目的:分析CD47靶向细胞治疗、小分子及miRNA的作用机制与临床前潜力。
方法细节:总结CD47-CAR-T、CAR-M的构建策略与体内外实验数据,以及小分子、miRNA下调CD47/SIRPα的研究结果。
结果解读:CD47-CAR-T细胞可直接杀伤CD47+肿瘤细胞,同时分泌IL-2增强免疫应答,在卵巢癌、肺癌等实体瘤的临床前研究中显示出抗肿瘤活性;CAR-M通过表达肿瘤特异性CAR,增强巨噬细胞对肿瘤细胞的吞噬作用,同时诱导M2向M1极化;小分子RRx-001可同时下调肿瘤细胞CD47与巨噬细胞SIRPα,目前处于小细胞肺癌的III期临床;miRNA如miR-200a、miR-708可直接靶向CD47的3’UTR,下调其表达,促进巨噬细胞吞噬肿瘤细胞。
产品关联:文献未提及具体实验产品,领域常规使用慢病毒载体构建CAR-T/M细胞,qRT-PCR检测miRNA表达。
4. Biomarker研究及发现成果
本文献中涉及的Biomarker主要包括CD47、钙网蛋白(CRT)及CD47受体占有率,其中CD47作为肿瘤免疫逃逸的核心分子,是治疗靶点与潜在预后Biomarker;CRT作为“吃我”信号,与CD47的平衡可预测治疗响应;CD47受体占有率是评估药物暴露的关键Biomarker。
Biomarker定位:CD47的筛选逻辑为“肿瘤组织表达分析→临床前功能验证→临床疗效关联”,CRT的验证逻辑为“免疫原性细胞死亡诱导→吞噬作用关联→临床响应预测”,CD47受体占有率的检测逻辑为“药物剂量爬坡→外周血细胞结合率检测→疗效与毒性关联”。研究过程详述:CD47在血液肿瘤(AML、非霍奇金淋巴瘤)与实体瘤(卵巢癌、结直肠癌)中高表达,通过免疫组化(IHC)、流式细胞术检测,其高表达与肿瘤不良预后相关;CRT在化疗、放疗等诱导的免疫原性细胞死亡中从内质网转移至细胞表面,通过与巨噬细胞表面的LRP1结合,传递“吃我”信号,与CD47的表达水平比值可预测CD47靶向治疗的响应;CD47受体占有率通过流式细胞术检测外周血红细胞与T细胞的CD47结合率,如Magrolimab在30mg/kg剂量下实现90%以上的受体占有率,Evorpacept在10-15mg/kg剂量下CD47受体占有率约90%,与疗效相关。
核心成果提炼:CD47高表达是肿瘤免疫逃逸的关键机制,靶向CD47的药物在血液肿瘤中显示出显著疗效,如Magrolimab联合阿扎胞苷在AML中的客观缓解率为65%(n=34,文献未明确提供P值),其中TP53突变患者的客观缓解率达71%(n=21,文献未明确提供P值);CRT表达可增强CD47靶向治疗的疗效,阿扎胞苷可诱导CRT表达,与Magrolimab联合发挥协同作用;CD47受体占有率是评估药物暴露的关键指标,高占有率与疗效相关,但也与血液毒性相关。推测:未来可通过联合CRT诱导剂、开发肿瘤选择性CD47靶向药物,提高实体瘤的治疗响应率,同时通过CD47与CRT的表达水平优化患者筛选策略。