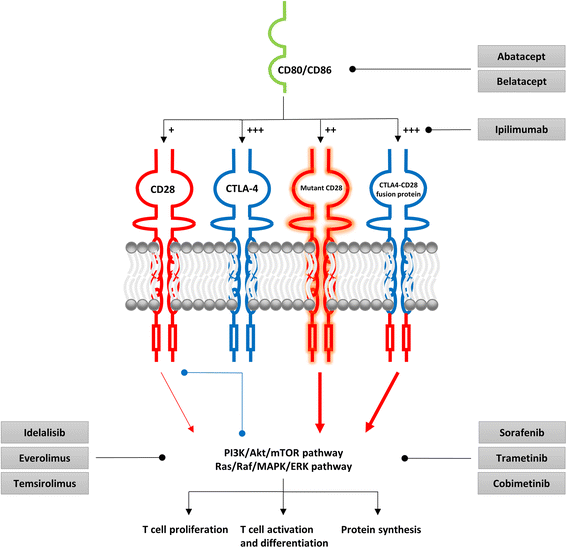
Angioimmunoblastic T-cell lymphoma (AITL) is one of the most common subtypes of peripheral T-cell lymphoma. Advances in understanding the mutational landscape of AITL have not resulted in improved prognosis nor consensus regarding optimal first-line and second-line treatment. The recently proposed multistep tumorigenesis model for AITL provides a theoretical framework of AITL oncogenesis. In this model, early mutations in epigenetic modifiers interact with late cooperative mutations to enable malignant transformation. Frequent mutations in epigenetic modifiers suggest that aberrant DNA methylation contributes to AITL oncogenesis. Several research groups have reported findings suggesting that inappropriate costimulation acts as a late cooperative mutation. Drugs targeting inappropriate costimulation have already been approved for the treatment of several malignancies or autoimmune diseases. Additionally, aberrant DNA methylation was recently shown to potentiate inappropriate costimulation in a subset of AITL cases. Therefore, drugs targeting inappropriate costimulation and hypomethylating agents might have synergistic effects. Both offer promising new therapeutic options in AITL treatment. This commentary summarizes the main findings on aberrant DNA methylation and inappropriate costimulation in AITL and proposes several already approved drugs for AITL treatment. Hopefully, these will contribute to improving the still dismal prognosis of AITL patients.
文献英文标题:Inappropriate costimulation and aberrant DNA methylation as therapeutic targets in angioimmunoblastic T-cell lymphoma;发表期刊:Biomarker Research;影响因子:未公开;研究领域:血管免疫母细胞性T细胞淋巴瘤(AITL)的分子机制与治疗靶点。