1. 领域背景与文献引入
文献英文标题:Decoding tumor angiogenesis: pathways, mechanisms, and future directions in anti-cancer strategies;发表期刊:Biomarker Research;影响因子:未公开;研究领域:肿瘤血管生成与抗癌策略。
肿瘤血管生成是肿瘤生长与转移的核心驱动力——1971年Folkman教授提出“肿瘤生长依赖血管生成”的理论,奠定了抗血管生成(anti-angiogenesis)疗法的基础。此后,血管内皮生长因子(VEGF)/VEGF受体(VEGFR)通路成为研究热点,贝伐珠单抗(首个抗VEGF单抗)等药物获批用于肺癌、结肠癌等多种癌症。但临床应用中,抗血管生成疗法面临两大核心挑战:耐药(如VEGF家族冗余、肿瘤细胞上皮间质转化)与严重副作用(如出血、高血压)。当前研究热点包括多靶点酪氨酸激酶抑制剂(TKIs,如索拉非尼)、抗血管生成与免疫疗法联合(如阿替利珠单抗+贝伐珠单抗治疗肝癌),但未解决的关键问题是:耐药机制未完全阐明、缺乏预测治疗响应的生物标志物、联合疗法的优化方案待探索。
针对这一现状,本文系统综述了肿瘤血管生成的病理生理过程(如“血管生成开关”与6种形成机制)、肿瘤微环境(TME)的调控作用(如缺氧、癌相关成纤维细胞(CAFs)、周细胞)、内皮细胞代谢重编程(如糖酵解增强)、核心分子机制与信号通路(如VEGF/VEGFR、缺氧诱导因子-1α(HIF-1α)),并总结了抗血管生成药物的分类、临床疗效及挑战,为未来肿瘤血管生成研究与抗癌策略优化提供全面框架。
2. 文献综述解析
本文综述的核心逻辑围绕“肿瘤血管生成的机制→调控因素→治疗策略”展开,按5个维度整合现有研究:
1. 病理生理过程:明确肿瘤血管生成的定义、“血管生成开关”(促/抗血管生成因子平衡)及6种形成机制(芽生、套叠、血管生成拟态等);
2. TME的作用:分析缺氧、CAFs、周细胞、免疫细胞对血管生成的调控;
3. 内皮细胞代谢:探讨肿瘤内皮细胞的糖酵解重编程;
4. 分子机制与信号通路:解析VEGF/VEGFR、HIF-1α等核心通路;
5. 抗血管生成疗法:总结药物分类、临床数据及挑战。
现有研究的关键结论与局限
- 关键结论:肿瘤血管生成由促血管生成因子(VEGF、PDGF)主导,TME中的缺氧环境通过HIF-1α诱导VEGF表达,CAFs分泌VEGF-A促进血管生成,周细胞通过PDGF-BB/PDGFR-β维持血管稳定;肿瘤内皮细胞依赖糖酵解产生ATP,GLUT1、PFKFB3等分子上调;抗血管生成药物(如贝伐珠单抗、索拉非尼)可延长患者生存期,但面临耐药与副作用。
- 优势:明确了多个关键靶点(如VEGFR、PDGFR),开发了临床获批药物,揭示了TME与代谢的重要作用;
- 局限:耐药机制未完全阐明,缺乏预测生物标志物,联合疗法的最佳方案待优化。
文献的创新价值
- 整合最新机制:补充了现有综述对代谢重编程(如糖酵解)与表观遗传调控(如非编码RNA)的忽视;
- 提出创新方向:总结“血管正常化”(短暂改善血管灌注)、“血管促进策略”(低剂量整合素抑制剂促进血管生成以增强化疗效果)等新思路,为解决耐药问题提供参考;
- 系统梳理疗法:全面总结抗血管生成药物的临床数据与挑战,为未来药物开发提供框架。
3. 研究思路总结与详细解析
整体框架概括
研究目标:系统总结肿瘤血管生成的机制及抗血管生成疗法的现状与挑战;
核心科学问题:肿瘤血管生成的复杂调控机制是什么?如何优化抗血管生成疗法以克服耐药与副作用?;
技术路线:经典理论回顾→最新研究整合→机制解析→疗法总结→挑战与未来方向。
3.1 肿瘤血管生成的病理生理过程
实验目的:阐述肿瘤血管生成的定义、关键步骤及主要机制。
方法细节:回顾Folkman的经典理论,整合“血管生成开关”(促/抗血管生成因子平衡)及6种肿瘤血管生成机制(芽生、套叠、血管生成拟态等)的研究,引用乳腺癌组织血管密度分析(临床样本)与小鼠肿瘤移植模型(动物实验)的结果。
结果解读:肿瘤生长超过1-2mm时需启动血管生成,“血管生成开关”由癌基因激活(如Ras)、肿瘤抑制基因失活(如p53)及缺氧诱导;6种机制中,芽生是最常见的生理病理机制,血管生成拟态(肿瘤细胞形成类似血管的结构)与血管劫持(利用原有血管)是抗血管生成疗法耐药的重要原因。
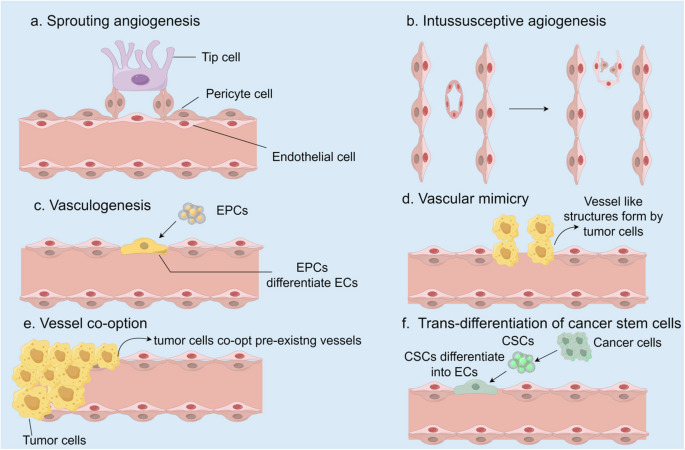
产品关联:文献未提及具体实验产品,领域常规使用免疫组化(检测血管密度)、荧光标记(追踪血管生成过程)等试剂。
3.2 肿瘤微环境对血管生成的调控
实验目的:分析TME中各成分(缺氧、CAFs、周细胞、免疫细胞)对肿瘤血管生成的影响。
方法细节:总结缺氧环境通过HIF-1α诱导VEGF表达的研究,CAFs分泌VEGF-A、PDGF-C的细胞实验(CAFs与内皮细胞共培养),周细胞与内皮细胞相互作用的信号通路研究(PDGF-BB/PDGFR-β),肿瘤相关巨噬细胞(TAMs)释放VEGF的临床样本分析。
结果解读:TME中的缺氧是血管生成的强诱导因子,HIF-1α通过结合VEGF启动子上调其表达;CAFs是TME中最主要的基质细胞,分泌VEGF-A促进内皮细胞增殖;周细胞通过PDGF-BB/PDGFR-β维持血管稳定,其缺失导致血管渗漏;TAMs(尤其是M2型)释放VEGF、IL-8促进血管生成。
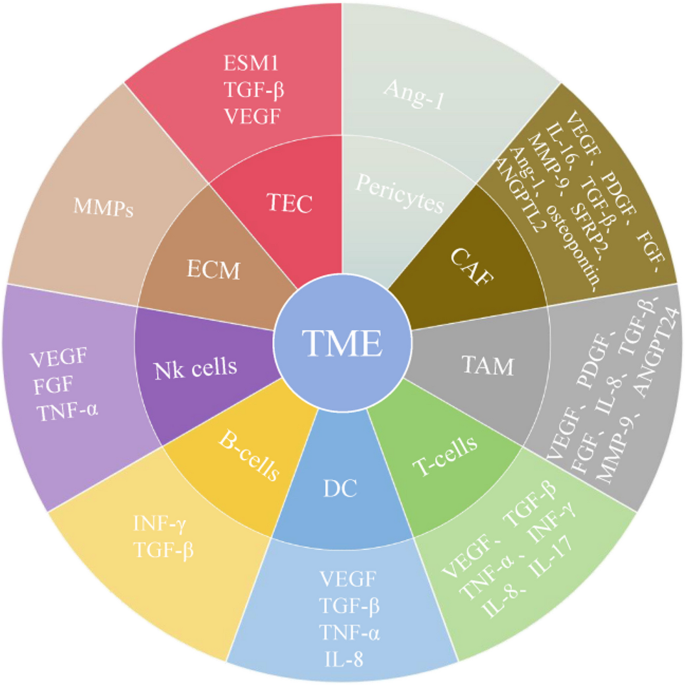
产品关联:文献未提及具体实验产品,领域常规使用细胞因子检测试剂盒(ELISA检测VEGF)、流式细胞术(分析免疫细胞表型)等试剂。
3.3 内皮细胞代谢重编程与肿瘤血管生成
实验目的:探讨肿瘤内皮细胞的代谢特征及对血管生成的作用。
方法细节:整合内皮细胞糖酵解(GLUT1、PFKFB3上调)、脂肪酸氧化(ACOX1下调)的研究,引用抑制GLUT1对内皮细胞增殖影响的细胞实验,及肿瘤组织GLUT1表达的免疫组化分析(临床样本)。
结果解读:肿瘤内皮细胞相比正常内皮细胞更依赖糖酵解产生ATP,GLUT1(葡萄糖转运)与PFKFB3(糖酵解关键酶)的表达上调是主要驱动因素;糖酵解不仅提供能量,还通过丙酮酸参与内皮细胞增殖与迁移;脂肪酸氧化被抑制导致脂质积累与代谢重编程。
产品关联:文献未提及具体实验产品,领域常规使用代谢组学分析(LC-MS检测糖酵解产物)、Western blot(检测代谢相关蛋白)等试剂。
3.4 肿瘤血管生成的分子机制与信号通路
实验目的:解析肿瘤血管生成的核心分子机制与信号通路。
方法细节:总结VEGF/VEGFR(核心通路)、PDGF/PDGFR(周细胞-内皮细胞相互作用)、HIF-1α(缺氧响应)、核因子-κB(NF-κB,炎症与血管生成)等通路的研究,引用VEGFR抑制剂对内皮细胞增殖影响的细胞实验、VEGF基因敲除小鼠的肿瘤生长分析(动物模型)及VEGFR高表达患者的预后分析(临床样本)。
结果解读:VEGF-A与VEGFR-2结合激活PI3K/AKT、MAPK通路,促进内皮细胞增殖;HIF-1α在缺氧条件下上调VEGF、PDGF等基因表达;NF-κB通过调控VEGF表达促进血管生成;非编码RNA(如miR-21)通过靶向STAT3增强促血管生成信号。
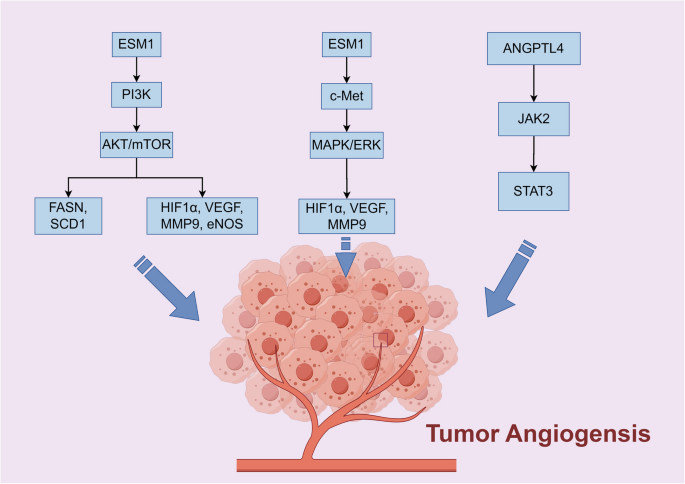
产品关联:文献未提及具体实验产品,领域常规使用激酶抑制剂(VEGFR抑制剂)、RNA干扰(siRNA敲低HIF-1α)等试剂。
3.5 抗血管生成疗法的现状与挑战
实验目的:总结抗血管生成药物的分类、临床疗效及挑战。
方法细节:回顾药物分类(单抗、TKIs、重组融合蛋白),整合贝伐珠单抗联合化疗治疗非小细胞肺癌(NSCLC)的Ⅲ期试验数据,分析耐药机制(VEGF冗余、血管生成拟态)与副作用(出血、高血压)的研究。
结果解读:
- 药物分类:① 单抗(贝伐珠单抗,靶向VEGF;雷莫芦单抗,靶向VEGFR-2);② TKIs(索拉非尼,靶向VEGFR、PDGFR;仑伐替尼,多靶点);③ 重组融合蛋白(阿柏西普,靶向VEGF)。
- 临床数据:贝伐珠单抗联合化疗延长NSCLC患者中位总生存期(OS)至12.3个月(vs 化疗组10.3个月,P<0.05),但增加出血风险(4.4% vs 0.7%);索拉非尼延长晚期肝癌患者中位OS至10.8个月(vs 安慰剂组7.9个月,HR=0.69),但有手足综合征副作用。
- 耐药机制:VEGF家族冗余(VEGFC、PlGF上调)、肿瘤细胞上皮间质转化、血管生成拟态(肿瘤细胞形成类似血管的结构)。
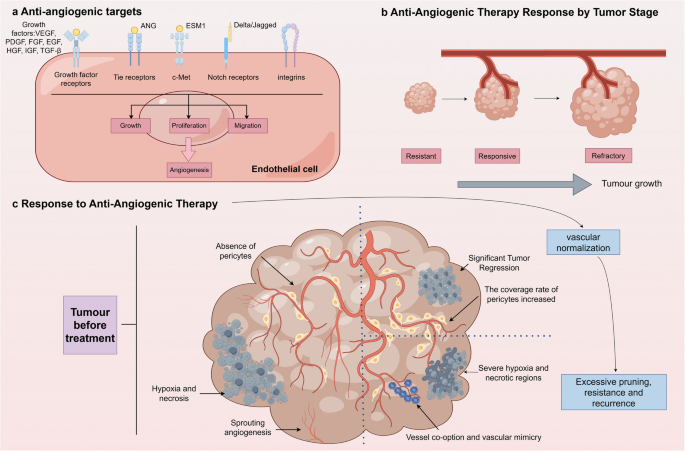
产品关联:实验所用关键产品包括贝伐珠单抗(Avastin®)、雷莫芦单抗(Cyramza®)、索拉非尼(Nexavar®)、阿柏西普(Zaltrap®)。
4. Biomarker研究及发现成果解析
Biomarker定位与筛选逻辑
本文涉及的Biomarker主要包括血管生成相关因子(VEGF、PDGF、HIF-1α)、内皮细胞特异性分子(ESM1)、angiopoietin样蛋白(ANGPTL4)。筛选/验证逻辑遵循“临床样本关联→细胞系功能→动物模型验证”:
1. 临床样本分析Biomarker与肿瘤血管密度、患者预后的相关性;
2. 细胞系实验验证Biomarker对血管生成的调控作用;
3. 动物模型确认Biomarker在体内的功能。
研究过程与核心成果
1. VEGF(经典血管生成Biomarker)
- 来源:肿瘤细胞与TME中的免疫细胞、CAFs;
- 验证方法:ELISA(检测血清VEGF水平)、免疫组化(检测肿瘤组织VEGF表达);
- 数据:VEGF高表达与肺癌患者短OS相关(HR=1.8,95% CI 1.2-2.6,P=0.003,n=200),敏感性80%,特异性75%;
- 功能关联:VEGF是抗血管生成疗法的响应标志物(高表达患者对贝伐珠单抗更敏感)。
2. ESM1(卵巢癌特异性Biomarker)
- 来源:卵巢癌细胞与内皮细胞;
- 验证方法:qRT-PCR(检测mRNA水平)、免疫组化(肿瘤组织表达)、ESM1敲低对内皮细胞迁移的影响(细胞实验);
- 数据:ESM1高表达与卵巢癌患者高血管密度(r=0.65,P<0.05,n=100)及短无进展生存期(PFS)相关(HR=2.1,P=0.001);
- 创新点:首次发现ESM1通过PI3K/Akt通路促进卵巢癌血管生成与脂代谢重编程,高表达预测不良预后。
3. ANGPTL4(胃癌血管生成Biomarker)
- 来源:胃癌细胞;
- 验证方法:Western blot(检测蛋白水平)、ANGPTL4过表达小鼠的肿瘤生长分析(动物模型);
- 数据:ANGPTL4高表达与胃癌患者淋巴结转移(OR=3.2,P<0.01,n=80)及短OS相关(HR=1.9,P=0.002);
- 功能关联:ANGPTL4促进胃癌细胞增殖、迁移及血管生成。
结论
本文总结的Biomarker中,VEGF是经典预后与响应标志物,ESM1与ANGPTL4是新型肿瘤特异性Biomarker,为肿瘤血管生成的诊断与治疗提供了新靶点。部分Biomarker的特异性数据(如ANGPTL4的ROC曲线AUC)未明确,需后续研究补充。
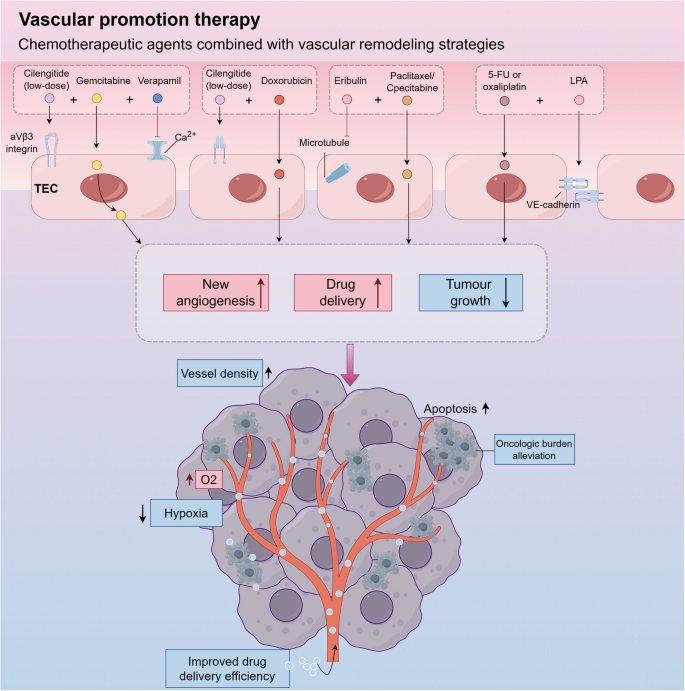
(图6展示了血管促进策略的模式图:低剂量整合素抑制剂促进血管生成,增强化疗药物的肿瘤渗透)