1. 领域背景与文献引入
文献英文标题:Brain dystrophin-glycoprotein complex: Persistent expression of β-dystroglycan, impaired oligomerization of Dp71 and up-regulation of utrophins in animal models of muscular dystrophy;发表期刊:BMC Cell Biology;影响因子:未公开;研究领域:杜氏肌营养不良症中枢神经系统病理机制。
杜氏肌营养不良症(DMD)是一种X连锁隐性遗传疾病,由抗肌萎缩蛋白(dystrophin)基因突变导致,主要病理特征为骨骼肌进行性坏死,患者多因呼吸或心脏衰竭死亡。领域共识:约30%的DMD患者伴随非进行性认知障碍,但其中枢神经系统的病理机制长期未被阐明。既往研究主要聚焦于肌肉组织中抗肌萎缩蛋白糖蛋白复合物(DGC)的功能,该复合物通过连接细胞外基质与细胞骨架维持肌膜稳定性,抗肌萎缩蛋白缺失会导致DGC解体,引发肌肉坏死。但脑内存在多种抗肌萎缩蛋白亚型(如神经元中的Dp427、血管胶质细胞中的Dp71),DGC组分在脑内的分布、功能及抗肌萎缩蛋白缺失后的变化规律尚不明确,且DMD患者与mdx小鼠模型中脑内β-肌营养不良蛋白聚糖(β-dystroglycan)的表达变化存在矛盾报道,这一领域空白限制了对DMD认知障碍分子机制的理解。本研究旨在利用两种抗肌萎缩蛋白缺失的小鼠模型(mdx缺失Dp427,mdx-3cv缺失所有抗肌萎缩蛋白亚型),系统分析脑内DGC组分的表达、定位及寡聚化状态,揭示脑内抗肌萎缩蛋白缺失后的病理代偿机制,为DMD认知障碍的干预提供分子基础。
2. 文献综述解析
作者以“组织特异性(肌肉vs中枢神经系统)-蛋白功能分类(抗肌萎缩蛋白亚型、DGC组分、营养蛋白utrophin)”为维度,系统梳理了DMD领域的研究现状,明确了脑内DGC研究的核心空白。
现有研究显示,肌肉组织中抗肌萎缩蛋白(Dp427)是DGC的核心组分,其缺失会导致DGC的α-肌营养不良蛋白聚糖(α-dystroglycan)、β-dystroglycan、α-肌聚多糖(α-sarcoglycan)等组分显著减少,肌膜稳定性丧失,最终引发肌肉坏死,这一机制在DMD患者及mdx小鼠模型中已得到充分验证。在中枢神经系统中,抗肌萎缩蛋白存在多种亚型:全长Dp427主要定位于神经元突触后膜,参与突触功能调控;短亚型Dp71主要分布于血管胶质细胞,与血脑屏障形成相关;营养蛋白utrophin作为抗肌萎缩蛋白的同源物,在正常脑内广泛分布,可能参与脑区特化维持。但现有研究存在两大局限性:一是未系统对比不同抗肌萎缩蛋白亚型缺失对脑内DGC组分的影响;二是对脑内抗肌萎缩蛋白缺失后的代偿机制(如utrophin的调控)缺乏深入解析,且DMD患者与mdx小鼠脑内β-dystroglycan的表达变化存在矛盾,无法为认知障碍提供统一的分子解释。本研究的创新价值在于,首次采用两种不同抗肌萎缩蛋白缺失的小鼠模型,从蛋白定位、表达水平、寡聚化状态三个层面系统解析脑内DGC的变化规律,明确了β-dystroglycan在脑内的特异性定位及抗肌萎缩蛋白缺失后的代偿机制,解决了既往研究中物种间的矛盾报道,为DMD中枢神经系统病理机制提供了新的视角。
3. 研究思路总结与详细解析
本研究以“抗肌萎缩蛋白缺失对脑内DGC的影响及代偿机制”为核心科学问题,采用“动物模型验证-蛋白定位分析-表达水平检测-寡聚化功能分析”的闭环技术路线,系统揭示了脑内抗肌萎缩蛋白缺失后的病理变化及代偿机制。
3.1 动物模型构建与验证
实验目的是建立并验证抗肌萎缩蛋白缺失的动物模型,对比不同亚型缺失对肌肉及脑内DGC的影响;方法细节是选取mdx小鼠(因exon23点突变缺失全长抗肌萎缩蛋白Dp427)和mdx-3cv小鼠(因exon65突变缺失所有抗肌萎缩蛋白亚型,包括脑内主要亚型Dp71),取胫骨前肌及前脑组织,液氮速冻后冷冻保存,通过免疫荧光及免疫印迹技术验证模型的有效性;结果解读:免疫荧光显示,两种模型小鼠肌肉组织中抗肌萎缩蛋白完全缺失,α-dystroglycan、β-dystroglycan、α-sarcoglycan等DGC组分的免疫荧光信号显著减弱,与既往研究结果一致,验证了模型的可靠性。
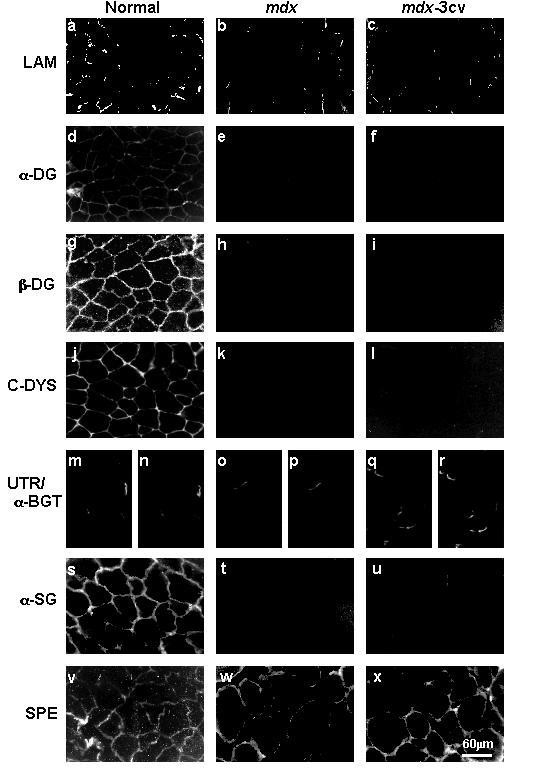
实验所用关键产品:Novocastra Laboratories的抗β-dystroglycan抗体(NCL-43)、抗α-sarcoglycan抗体(NCL-a-SARC),Upstate Biotechnology的抗α-dystroglycan抗体(VIA4₁)等。
3.2 脑内β-dystroglycan的细胞定位分析
实验目的是明确β-dystroglycan在正常小鼠脑内的细胞分布,为后续分析其功能奠定基础;方法细节是采用免疫荧光双标技术,将前脑冷冻切片分别与β-dystroglycan抗体及细胞特异性标志物抗体(神经丝蛋白标记神经元、胶质纤维酸性蛋白标记星形胶质细胞、血管性血友病因子vWF标记内皮细胞)共孵育,通过荧光显微镜观察共定位情况;结果解读:β-dystroglycan与神经元标志物无共定位,与星形胶质细胞标志物部分共定位,与内皮细胞标志物vWF高度共定位,表明β-dystroglycan主要定位于脑内血管胶质界面(血脑屏障的星形胶质细胞足突),而非神经元。
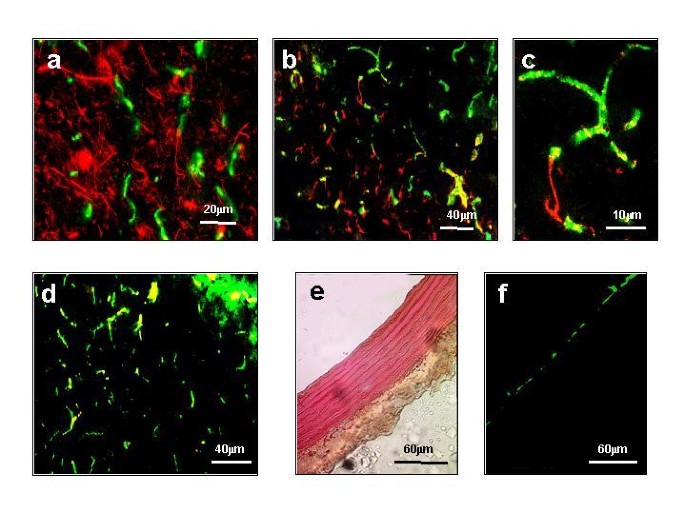
实验所用关键产品:Sigma Chemical Company的抗vWF抗体、抗胶质纤维酸性蛋白抗体,Novocastra Laboratories的抗β-dystroglycan抗体(NCL-43)等。
3.3 脑内DGC组分的表达水平检测
实验目的是对比抗肌萎缩蛋白缺失后,肌肉与脑内DGC组分的表达变化差异;方法细节是提取正常小鼠、mdx小鼠、mdx-3cv小鼠的肌肉及脑微粒体膜蛋白,采用免疫印迹技术检测laminin、α-dystroglycan、β-dystroglycan、抗肌萎缩蛋白亚型、utrophin亚型的表达水平;结果解读:肌肉组织中,两种模型小鼠的α-dystroglycan、β-dystroglycan、α-sarcoglycan表达均显著减少,与既往研究一致;而脑内组织中,mdx及mdx-3cv小鼠的β-dystroglycan表达不受影响,α-dystroglycan及laminin表达减少,utrophin亚型Up116和Up71在mdx-3cv小鼠脑内显著上调,全长utrophin表达无变化。
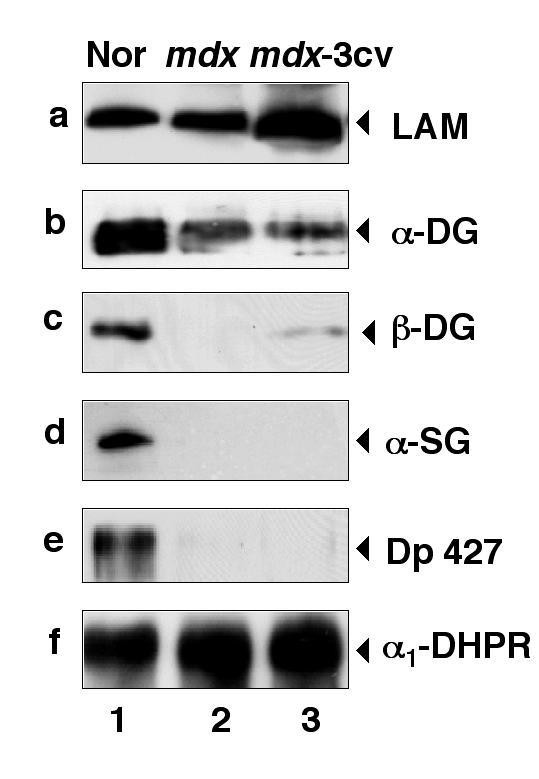
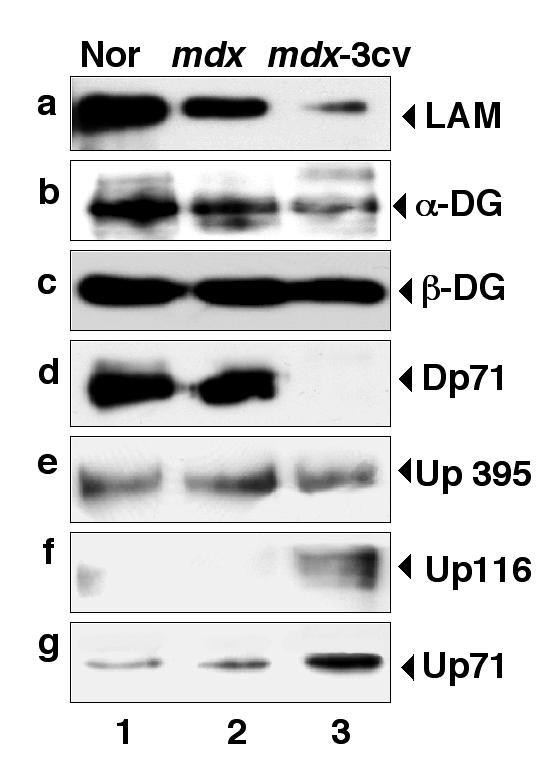
实验所用关键产品:Novocastra Laboratories的抗Dp71抗体、抗utrophin抗体,Upstate Biotechnology的抗Na⁺/K⁺-ATPase α亚基抗体(内参)等。
3.4 Dp71寡聚化状态的功能分析
实验目的是检测脑内主要抗肌萎缩蛋白亚型Dp71的寡聚化状态,明确抗肌萎缩蛋白缺失对其功能的影响;方法细节是采用化学交联技术,用亲水性交联剂BS³处理正常小鼠、mdx小鼠的脑微粒体膜蛋白,通过免疫印迹检测Dp71的迁移率变化,分析其寡聚化状态;结果解读:正常小鼠脑内Dp71经交联后形成高分子量寡聚体,而mdx小鼠脑内Dp71主要以单体形式存在,表明抗肌萎缩蛋白缺失导致Dp71的寡聚化受损,这可能影响其与DGC组分的结合及功能发挥。
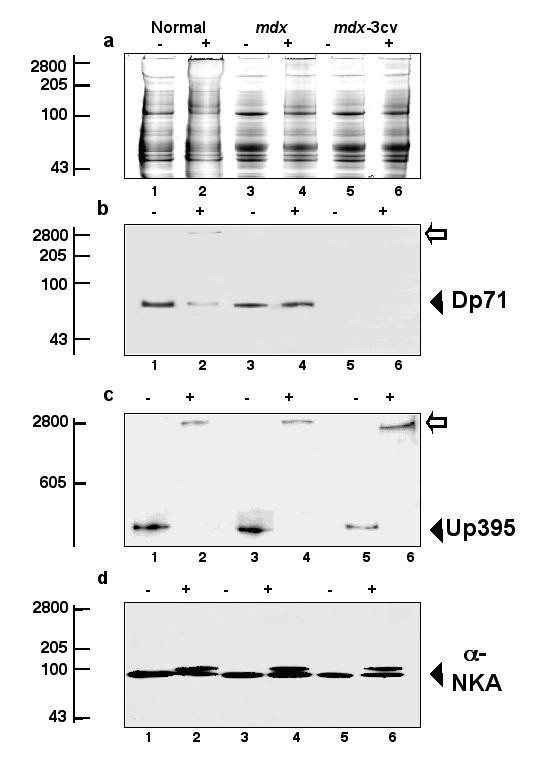
实验所用关键产品:Pierce & Warriner的化学交联剂BS³,Novocastra Laboratories的抗Dp71抗体等。
4. Biomarker研究及发现成果解析
本研究鉴定了三类与DMD中枢神经系统病理相关的Biomarker:β-dystroglycan的细胞定位、Dp71的寡聚化状态、utrophin亚型的上调水平,其筛选验证逻辑为“前期研究提示-细胞/动物模型验证-功能关联分析”。
Biomarker定位方面,β-dystroglycan作为脑内DGC的关键组分,其定位Biomarker的筛选逻辑为:前期研究提示其在脑内表达,但定位不明确→通过免疫荧光双标验证其在血管胶质界面的特异性定位→明确其与血脑屏障功能的关联;Dp71的寡聚化状态作为功能Biomarker,筛选逻辑为:Dp71是脑内主要抗肌萎缩蛋白亚型→化学交联实验验证其在抗肌萎缩蛋白缺失后寡聚化受损→关联其与DGC功能的丧失;utrophin亚型Up116/Up71作为代偿Biomarker,筛选逻辑为:utrophin是抗肌萎缩蛋白的同源物→免疫印迹验证其在抗肌萎缩蛋白缺失后上调→关联其对β-dystroglycan膜定位的维持作用。
研究过程详述:β-dystroglycan的定位验证采用免疫荧光双标法,以vWF为内皮细胞标志物,结果显示二者高度共定位(文献未明确提供样本量,基于荧光图像趋势推测);Dp71的寡聚化状态采用化学交联法,正常脑内Dp71形成高分子量复合物,mdx脑内无此复合物(文献未明确提供样本量,基于免疫印迹条带趋势推测);utrophin亚型的表达采用免疫印迹法,mdx-3cv小鼠脑内Up116/Up71的条带灰度值显著高于正常小鼠(文献未明确提供定量数据,基于条带强度趋势推测)。
核心成果提炼:β-dystroglycan在脑内血管胶质界面持续表达,不受抗肌萎缩蛋白缺失影响,这一发现解决了既往研究中DMD患者与mdx小鼠的矛盾报道;Dp71寡聚化受损是DMD中枢神经系统病理的关键功能Biomarker,其寡聚化异常可能导致DGC功能丧失,进而影响血脑屏障稳定性或突触功能;utrophin亚型Up116/Up71的上调是脑内重要的代偿机制,可能通过结合β-dystroglycan维持其膜定位,部分代偿抗肌萎缩蛋白的功能。本研究首次揭示了脑内抗肌萎缩蛋白缺失后的代偿机制,为DMD认知障碍的Biomarker开发及干预靶点筛选提供了新的方向,其中Dp71寡聚化状态的创新性在于,首次将抗肌萎缩蛋白的寡聚化状态与脑功能异常关联,为DMD中枢神经系统病理机制的研究开辟了新视角。