1. 领域背景与文献引入
文献英文标题:Activation of GPR40 attenuates chronic inflammation induced impact on pancreatic β-cells health and function;发表期刊:BMC Cell Biology;影响因子:未公开;研究领域:糖尿病与胰腺β细胞生物学。
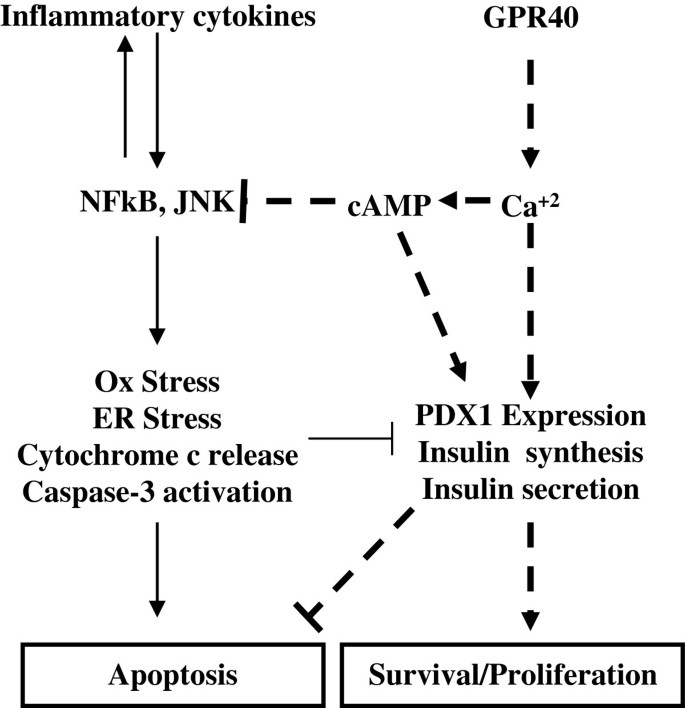
糖尿病(1型和2型)的核心发病机制是胰腺β细胞绝对量减少及功能障碍,其中慢性炎症介导的β细胞凋亡是导致β细胞量下降的关键原因。领域共识:2000年后,研究逐步明确炎症因子(如TNFα、IL1β)通过激活JNK、NFκB通路诱导β细胞氧化应激、内质网应激,最终引发凋亡;2003年GPR40(游离脂肪酸受体1)被发现高表达于β细胞,参与脂肪酸介导的胰岛素分泌调控,成为糖尿病治疗的潜在靶点。当前研究热点聚焦于寻找能同时保护β细胞存活与功能的靶向策略,但未解决的核心问题是GPR40激活在抗炎及抗β细胞凋亡中的具体作用机制尚未系统阐明,缺乏将GPR40与炎症-凋亡信号轴关联的深入研究。针对这一研究空白,本研究旨在探究GPR40特异性激动剂CNX-011-67对慢性炎症诱导的β细胞损伤的保护作用及下游信号通路,为糖尿病治疗提供新的靶点与理论依据。
2. 文献综述解析
作者对领域内现有研究的分类维度为β细胞损伤机制(炎症应激、凋亡通路)与保护策略(如GLP-1类似物靶向治疗)两类。现有研究的关键结论包括:慢性炎症通过激活JNK、NFκB通路上调促炎因子表达,形成正反馈循环加重β细胞损伤;GLP-1类似物可通过提升cAMP水平抑制β细胞凋亡并促进胰岛素分泌。技术方法优势在于多采用细胞系与原代胰岛模型结合的方式,能较好模拟体内β细胞的生理环境;局限性则体现在GPR40相关研究多聚焦于胰岛素分泌调控,对其抗炎及抗凋亡的作用机制研究不足,缺乏系统的信号通路解析,且未明确GPR40激活与β细胞存活信号的直接关联。
通过对比现有研究的未解决问题,本研究的创新价值凸显:首次系统阐明GPR40激活通过PLC-CaMKII-钙调磷酸酶-cAMP信号通路,同时抑制炎症介导的β细胞凋亡、恢复胰岛素合成与分泌,填补了GPR40在β细胞抗炎保护机制研究中的空白,为GPR40激动剂的临床转化提供了直接的实验依据。
3. 研究思路总结与详细解析
本研究的整体框架为:以“慢性炎症诱导β细胞凋亡与功能障碍”为核心科学问题,设定“验证GPR40激活的保护作用及解析下游机制”为研究目标,采用“GPR40特异性验证→炎症模型构建→多维度指标检测→信号通路解析→功能恢复验证”的闭环技术路线,结合细胞系与原代胰岛模型,系统揭示GPR40激活的生物学效应与分子机制。
3.1 GPR40激活的特异性验证
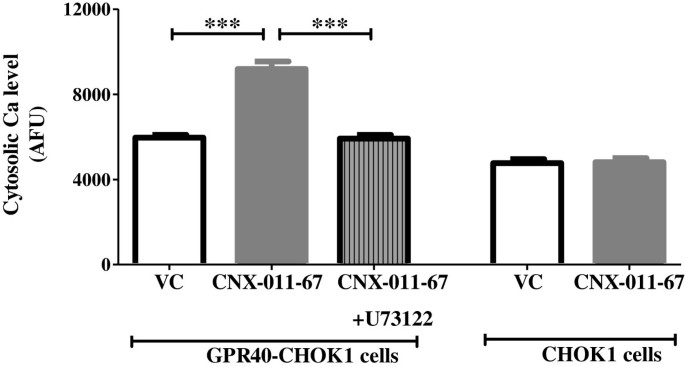
实验目的是确认CNX-011-67作为GPR40激动剂的特异性及信号通路依赖性。方法细节为采用过表达小鼠GPR40的CHOK1细胞与正常CHOK1细胞,给予1μM CNX-011-67处理,同时用PLC抑制剂U73122预处理,通过Fluo-4-AM荧光探针检测细胞质钙通量变化。结果解读:过表达GPR40的细胞中钙通量显著升高(9187 AFU vs 对照组5967 AFU,n=4,P<0.001),U73122处理后钙通量降至5921 AFU,正常CHOK1细胞无显著变化,说明CNX-011-67可特异性激活GPR40且依赖PLC通路。产品关联:文献未提及具体实验产品,领域常规使用钙荧光探针(如Fluo-4-AM)、PLC抑制剂等试剂。
3.2 炎症模型构建与GPR40抗炎作用验证
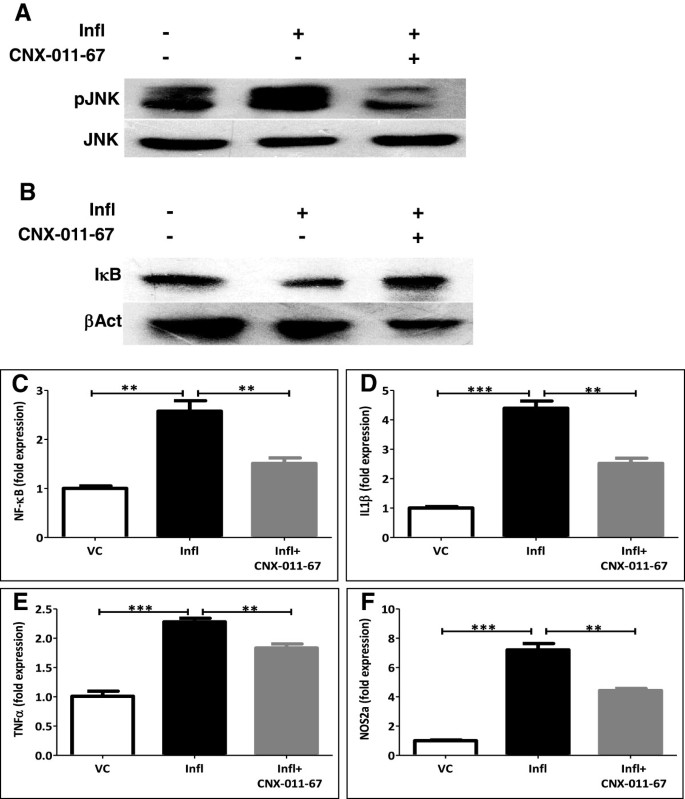
实验目的是验证GPR40激活对炎症信号通路的抑制作用。方法细节为用NIT1胰岛素瘤细胞和大鼠胰岛,给予TNFα和IL1β(各10ng/ml)处理72h构建慢性炎症模型,同时加入1μM CNX-011-67,通过蛋白质免疫印迹(WB)检测JNK磷酸化和IκB水平,qRT-PCR检测NF-κB、IL1β、TNFα、NOS2a基因表达。结果解读:炎症模型中JNK磷酸化升高4.4倍,IκB水平降低60%(n=4,P<0.001),NF-κB、IL1β、TNFα、NOS2a基因表达分别上调2.6、4.3、2.2、7.2倍(n=4,P<0.01或P<0.001);GPR40激活后JNK磷酸化降至1.5倍,IκB水平升至1.3倍,上述基因表达均显著降低,说明GPR40激活可有效抑制炎症信号通路的激活与促炎因子的表达。产品关联:实验所用关键产品:Cell Signaling Technology的pAKT、AKT、pJNK、JNK、IkB和β-actin抗体。
3.3 GPR40激活对细胞应激的影响
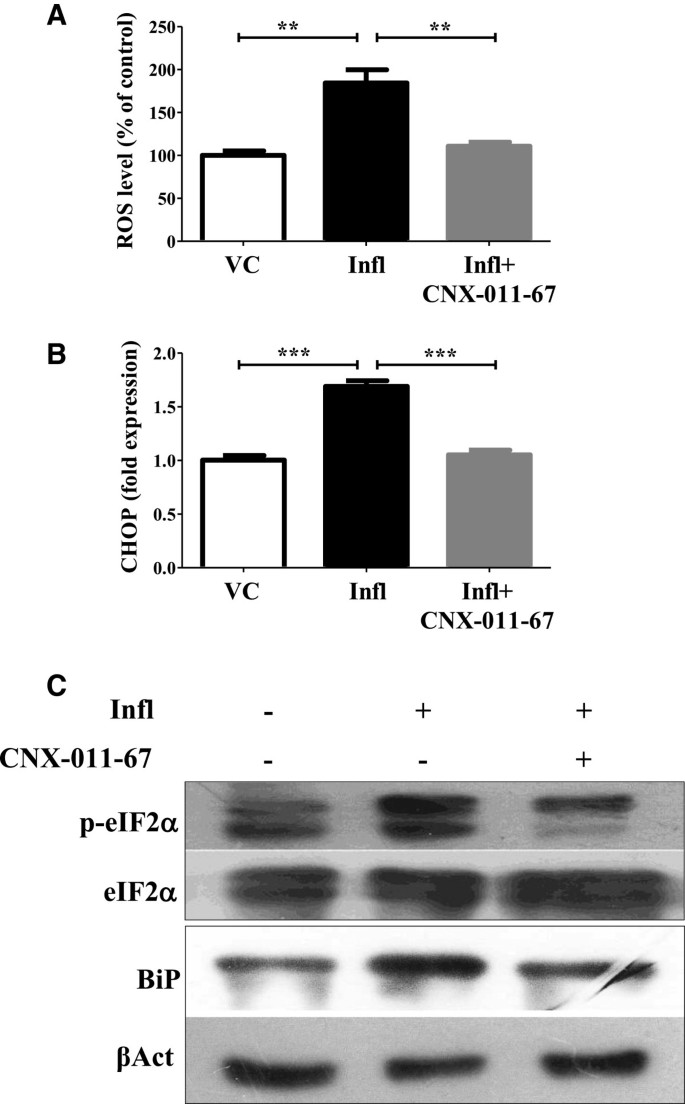
实验目的是检测GPR40激活对炎症诱导的氧化应激和内质网(ER)应激的调控作用。方法细节为NIT1细胞经炎症和GPR40激活处理72h后,用DCF荧光检测ROS水平,qRT-PCR检测CHOP基因表达,WB检测BiP和eIF2α磷酸化水平。结果解读:炎症组ROS水平升高1.8倍(n=4,P<0.01),CHOP基因表达上调1.6倍,BiP水平和eIF2α磷酸化分别升高1.4和1.2倍;GPR40激活后ROS水平恢复至正常,CHOP基因表达降至0.6倍,BiP水平和eIF2α磷酸化分别降至0.6和0.7倍,说明GPR40激活可显著降低炎症诱导的氧化应激与内质网应激。产品关联:文献未提及具体实验产品,领域常规使用DCFH-DA荧光探针、ER应激相关抗体等试剂。
3.4 GPR40激活对β细胞凋亡的抑制作用
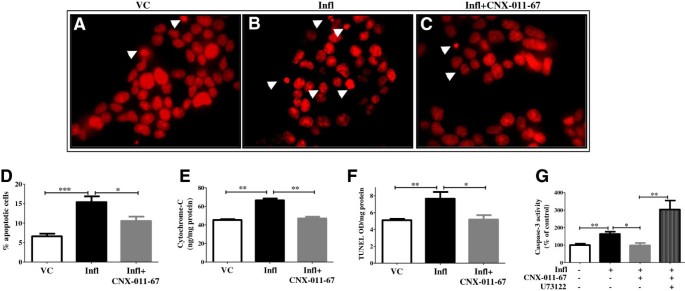
实验目的是验证GPR40激活对炎症诱导的β细胞凋亡的保护作用。方法细节为NIT1细胞和大鼠胰岛经处理后,通过核碎裂实验(碘化丙啶染色)、细胞色素c ELISA、TUNEL实验、Caspase-3活性检测多维度评估凋亡水平。结果解读:炎症组凋亡核比例从6.6%升至15.4%(n=4,P<0.001),细胞色素c水平、TUNEL阳性率、Caspase-3活性均显著升高;GPR40激活后凋亡核比例降至10.5%(n=4,P<0.05),其余凋亡指标也显著降低,且该保护作用被PLC抑制剂U73122逆转,说明GPR40激活可特异性抑制炎症诱导的β细胞凋亡。产品关联:实验所用关键产品:Invitrogen的Caspase-3底物Ac-DEVD-R110,Millipore的ApopTag® Peroxidase Apoptosis Detection Kit。
3.5 GPR40抗凋亡的信号通路解析
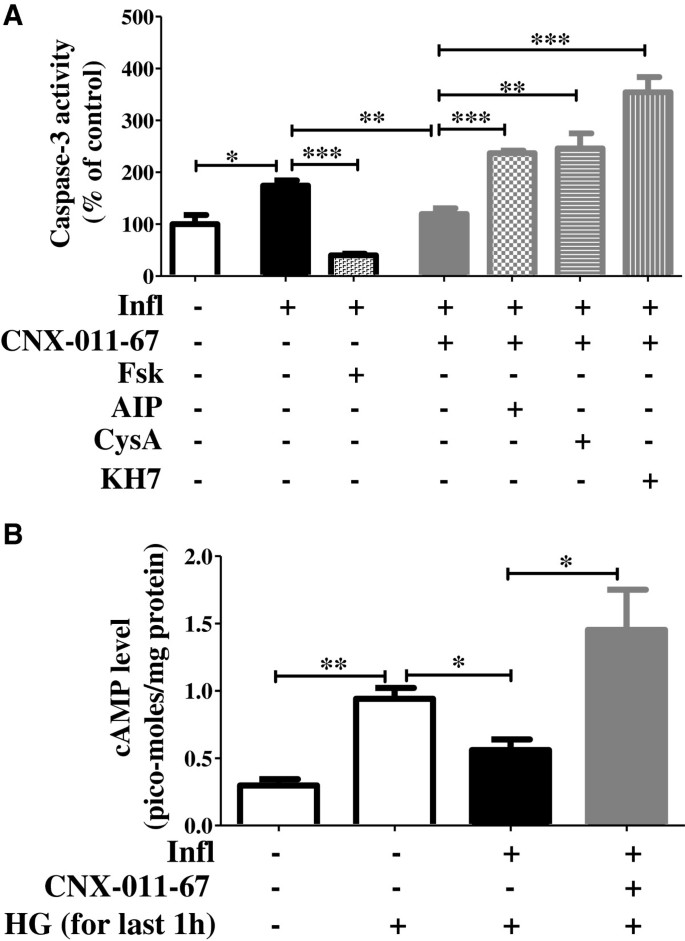
实验目的是解析GPR40激活抑制β细胞凋亡的下游信号通路。方法细节为用CaMKII抑制剂AIP、钙调磷酸酶抑制剂环孢素A、可溶性腺苷酸环化酶(ADCY)抑制剂KH7预处理NIT1细胞,检测Caspase-3活性变化;同时采用酶联免疫法检测细胞内cAMP水平。结果解读:GPR40激活使Caspase-3活性从炎症组的174%降至120%(n=4,P<0.01),抑制CaMKII或钙调磷酸酶后活性升至237%和246%,抑制ADCY后升至354%;炎症组高糖刺激下的cAMP水平从0.94pmol/mg蛋白降至0.56pmol/mg蛋白,GPR40激活后恢复至1.45pmol/mg蛋白(n=4,P<0.001),说明GPR40通过CaMKII-钙调磷酸酶-cAMP通路抑制β细胞凋亡。产品关联:文献未提及具体实验产品,领域常规使用信号通路抑制剂、cAMP检测试剂盒等试剂。
3.6 GPR40激活对β细胞存活信号的影响
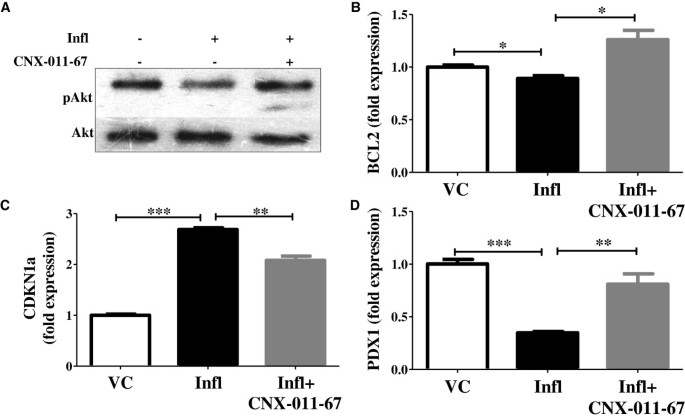
实验目的是检测GPR40激活对β细胞存活相关信号通路的调控作用。方法细节为通过WB检测Akt磷酸化水平,qRT-PCR检测BCL2、CDKN1a、PDX1基因表达。结果解读:炎症组Akt磷酸化降至对照组的0.54倍,BCL2和PDX1表达分别降至0.89和0.35倍,CDKN1a表达升至2.7倍(n=4,P<0.05或P<0.01);GPR40激活后Akt磷酸化升至1.51倍,BCL2和PDX1表达分别升至1.26和0.81倍,CDKN1a表达降至2.1倍,说明GPR40激活可增强β细胞存活信号,促进β细胞表型维持。产品关联:实验所用关键产品:Cell Signaling Technology的pAKT和AKT抗体。
3.7 GPR40激活对胰岛素合成与分泌的影响
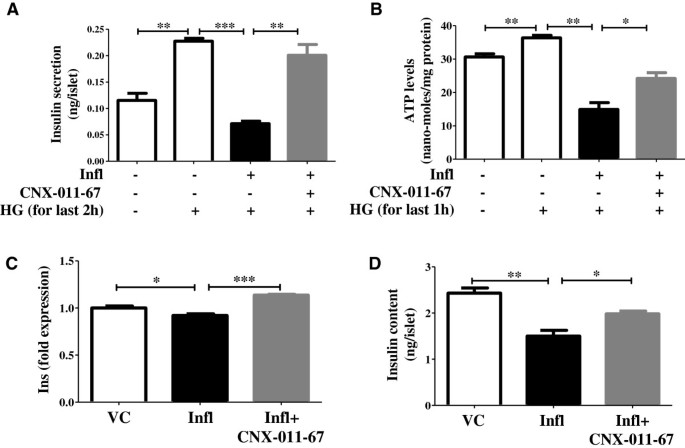
实验目的是验证GPR40激活对炎症诱导的β细胞功能障碍的恢复作用。方法细节为大鼠胰岛经处理后,检测高糖刺激的胰岛素分泌(GSIS)、细胞内胰岛素含量、ATP水平及胰岛素基因表达。结果解读:炎症组高糖刺激的胰岛素分泌从0.22ng/胰岛降至0.07ng/胰岛,细胞内胰岛素含量从2.4ng/胰岛降至1.5ng/胰岛(n=4,P<0.001),ATP水平从36nmol/mg蛋白降至15nmol/mg蛋白,胰岛素基因表达降至0.92倍;GPR40激活后上述指标均显著恢复,说明GPR40激活可有效改善β细胞的胰岛素合成与分泌功能。产品关联:实验所用关键产品:Mercodia的胰岛素ELISA试剂盒,Invitrogen的ATP测定试剂盒。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究涉及的Biomarker涵盖三类:炎症相关Biomarker(JNK磷酸化、IκB、NF-κB、IL1β、TNFα、NOS2a)、凋亡相关Biomarker(Caspase-3活性、细胞色素c、TUNEL阳性率)、存活与功能相关Biomarker(pAkt、BCL2、PDX1、胰岛素分泌量、胰岛素含量)。筛选与验证逻辑为“细胞模型初筛→原代胰岛验证→信号通路抑制剂反向验证”的三级体系,确保Biomarker的特异性与功能性。
研究过程详述
Biomarker来源为NIT1细胞与大鼠胰岛样本,验证方法包括蛋白质免疫印迹、qRT-PCR、ELISA、荧光检测等多种技术。特异性与敏感性数据显示:GPR40激活对JNK磷酸化的抑制率达65.9%(从4.4倍降至1.5倍,n=4,P<0.001),对Caspase-3活性的抑制率达31.0%(从174%降至120%,n=4,P<0.01);高糖刺激下,GPR40激活使胰岛素分泌量提升185.7%(从0.07ng/胰岛升至0.20ng/胰岛,n=4,P<0.001)。
核心成果提炼
这些Biomarker的功能关联在于,GPR40激活通过调控炎症-凋亡-存活信号轴,实现对β细胞的全方位保护:其中pAkt、BCL2作为存活Biomarker,GPR40激活后pAkt水平显著升高(n=4,P<0.01),BCL2表达上调(n=4,P<0.05),直接反映β细胞存活能力提升;PDX1作为β细胞表型维持Biomarker,其表达恢复可促进胰岛素合成。本研究的创新性在于首次揭示GPR40激活通过PLC-CaMKII-钙调磷酸酶-cAMP通路同步调控多类Biomarker,为糖尿病治疗提供了新的靶点组合与疗效评估指标,且所有结论均有明确的统计学结果支持(P<0.05至P<0.001)。