1. 领域背景与文献引入
文献英文标题:PKP1 promotes lung cancer by modulating energy metabolism through stabilization of PFKP;发表期刊:Biomarker Research;影响因子:未公开;研究领域:肺癌(肺鳞状细胞癌)代谢调控与靶向治疗。
肺癌是全球癌症相关死亡的首要原因,2022年全球癌症统计数据显示其死亡率占所有癌症的18%以上,其中非小细胞肺癌(NSCLC)占85%,肺鳞状细胞癌(LUSC)是NSCLC的主要亚型之一。与肺腺癌(LUAD)相比,LUSC的驱动基因(如EGFR、KRAS)突变率极低(不足5%),缺乏针对性的靶向治疗药物,临床治疗仍以化疗、放疗为主,疗效有限且易产生耐药性。
桥粒蛋白plakophilin-1(PKP1)是细胞黏附结构的重要组件,传统观点认为其通过维持上皮细胞结构完整性发挥肿瘤抑制作用。然而,近年转录组学研究发现,PKP1是LUSC中差异表达最显著的基因之一,其高表达与患者不良预后相关,这与传统认知存在矛盾。代谢重编程是癌症的核心hallmark之一,尽管Warburg效应(有氧糖酵解)是经典的代谢表型,但肿瘤细胞的代谢具有异质性,线粒体功能的维持对肿瘤增殖同样关键。然而,PKP1在LUSC代谢重编程中的功能及分子机制尚未阐明,这一空白限制了LUSC代谢靶向治疗的发展。
本研究旨在解析PKP1在LUSC中的生物学功能,揭示其调控能量代谢的分子机制,为LUSC的靶向治疗提供新的理论依据和潜在靶点。
2. 文献综述解析
文献综述以“PKP1的传统功能与LUSC中的异常表达矛盾”为核心逻辑,系统评述了LUSC的治疗现状、代谢重编程的重要性及PKP1的研究进展。
首先,作者指出LUSC的治疗困境:由于EGFR、KRAS等常见驱动基因的突变率极低,现有靶向治疗药物对LUSC疗效甚微,亟需挖掘新的治疗靶点。其次,代谢重编程是癌症细胞的重要特征,尽管Warburg效应是经典的代谢表型,但越来越多的研究表明,肿瘤细胞的代谢具有异质性,线粒体功能的维持对肿瘤增殖同样关键,代谢重编程已成为癌症治疗的重要靶点。接着,作者综述了PKP1的已知功能:作为桥粒的核心组件,PKP1通过维持细胞间连接发挥肿瘤抑制作用;近年研究发现,PKP1可通过调控c-Myc的翻译促进LUSC增殖,但关于其在代谢中的作用尚未报道。
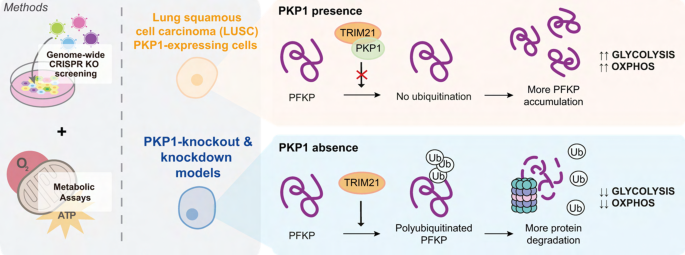
现有研究的局限性在于,未明确PKP1在LUSC代谢重编程中的角色,以及其高表达与肿瘤进展的分子联系。本研究的创新之处在于,首次将PKP1与LUSC的代谢重编程关联,揭示了PKP1通过稳定糖酵解限速酶PFKP调控线粒体功能和糖酵解的机制,为LUSC的代谢靶向治疗提供了新的靶点组合(PKP1-PFKP轴)。
3. 研究思路总结与详细解析
本研究以“解析PKP1在LUSC中的代谢调控机制”为核心目标,围绕“PKP1如何调控LUSC的能量代谢”这一科学问题,采用“CRISPR筛选→代谢表型验证→分子机制解析→功能验证”的闭环技术路线,逐步揭示PKP1调控能量代谢的分子通路。
3.1 CRISPR knockout筛选鉴定代谢依赖
实验目的:通过全基因组CRISPR knockout筛选,鉴定PKP1缺陷LUSC细胞的代谢依赖基因,揭示PKP1调控代谢的潜在途径。
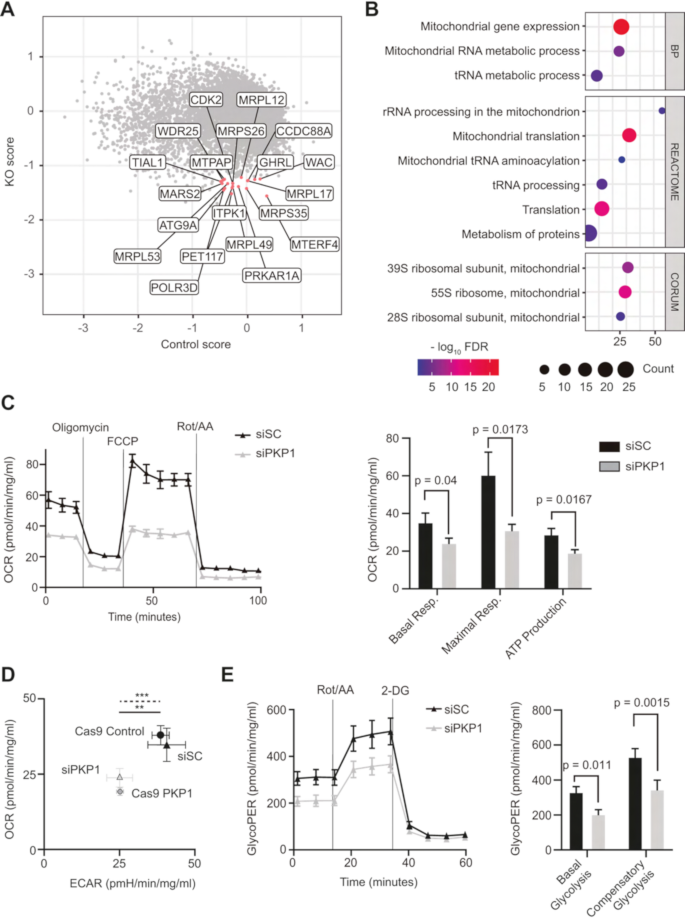
方法细节:在内在性表达PKP1的LUSC细胞系SK-MES-1中,利用CRISPR-Cas9技术构建两个PKP1双等位基因敲除(KO)克隆,同时以空载体转染细胞作为对照;采用全基因组sgRNA文库(覆盖约18000个基因,每个基因设计4条sgRNA)进行CRISPR knockout筛选,分别在筛选基线(第0天,sgRNA质粒库)和终点(第21天,细胞存活后)收集细胞,提取基因组DNA后进行高通量测序,分析sgRNA的丰度变化;计算每个基因的必需性评分(KO细胞与对照细胞的sgRNA丰度差异),筛选KO细胞中必需性评分最低(top 100)且对照细胞中评分在-0.5至0.5之间的基因;利用DAVID数据库对top 100基因进行功能富集分析,包括GO Biological Process、REACTOME通路和CORUM复合物。
结果解读:筛选结果显示,top hits主要为线粒体核糖体结构蛋白(如MRPS35、MRPS26、MRPL49、MRPL53)和线粒体功能相关蛋白(如PET117、MTERF4),这些基因在PKP1 KO细胞中的必需性评分显著低于对照细胞;功能富集分析表明,这些基因显著富集于线粒体相关通路(如“线粒体核糖体小亚基组装”“线粒体翻译”“氧化磷酸化”),-log₁₀ FDR值均大于2,提示PKP1缺陷的LUSC细胞对线粒体代谢存在强依赖,PKP1可能通过调控线粒体功能影响细胞代谢。
产品关联:文献未提及具体实验产品,领域常规使用Addgene的全基因组sgRNA文库、Illumina NovaSeq 6000测序平台进行sgRNA丰度分析,利用DAVID或Metascape进行功能富集分析。
3.2 代谢表型验证(线粒体功能与糖酵解)
实验目的:验证PKP1 depletion对LUSC细胞线粒体功能及糖酵解的影响,明确其代谢表型变化。
方法细节:构建两种PKP1缺陷模型:1)CRISPR-Cas9介导的PKP1双等位基因敲除(KO);2)siRNA介导的PKP1敲低(KD),以野生型SK-MES-1细胞或siRNA对照(siSC)为对照;使用Seahorse Bioscience XF96分析仪检测氧消耗率(OCR),依次加入寡霉素(抑制ATP合酶)、FCCP(解偶联剂)、鱼藤酮/抗霉素A(抑制电子传递链),计算基础呼吸、最大呼吸、ATP生成;检测细胞外酸化率(ECAR)和糖酵解质子外流率(glycoPER),依次加入鱼藤酮/抗霉素A(抑制线粒体呼吸)、2-脱氧葡萄糖(抑制糖酵解),计算基础糖酵解和代偿性糖酵解;通过免疫印迹(Western blot)检测线粒体标志物TOM20的表达,评估线粒体丰度。
结果解读:PKP1 KO和KD模型中,OCR(基础呼吸、最大呼吸及ATP生成)均显著低于对照细胞(n=3,P<0.01),表明线粒体功能受损;ECAR和glycoPER也显著降低(n=3,P<0.05),提示糖酵解活性下降;TOM20表达无显著变化(n=3,P>0.05),说明线粒体丰度未受影响,PKP1主要调控线粒体功能而非数量。能量map分析显示,PKP1表达细胞呈现高OCR和高ECAR的代谢活跃表型,而PKP1缺陷细胞代谢活性显著降低,处于“代谢静止”状态。
产品关联:实验所用关键产品:Seahorse Bioscience XF96分析仪(检测OCR/ECAR)、TOM20抗体(Cell Signaling Technology #42406,用于检测线粒体丰度);文献未提及具体品牌,领域常规使用上述产品。
3.3 PKP1对PFKP表达及稳定性的调控
实验目的:探究PKP1调控代谢的下游分子,明确其对糖酵解限速酶的影响及机制。
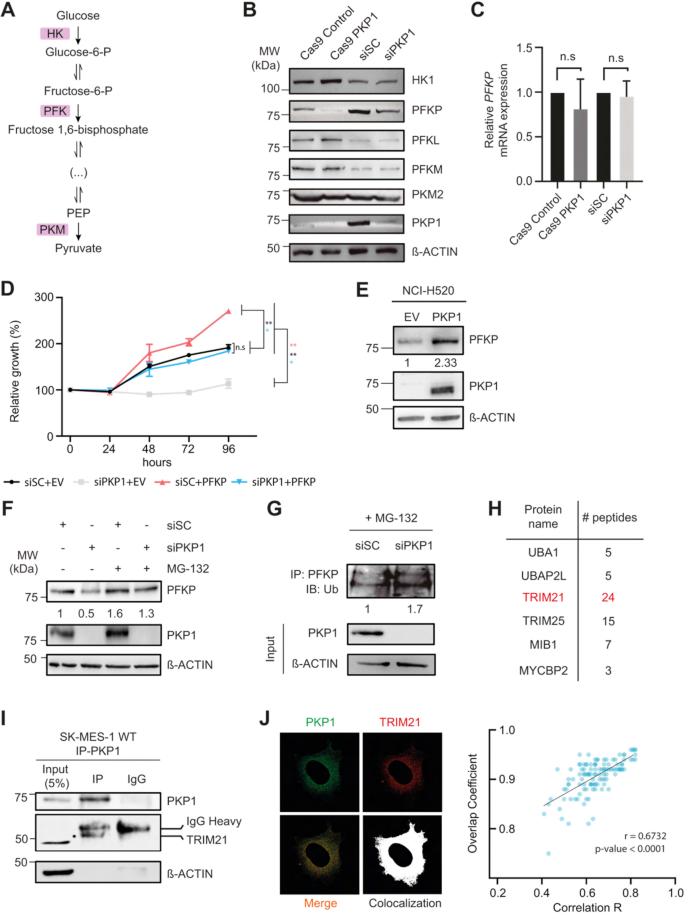
方法细节:通过qRT-PCR检测PKP1 KO/KD或过表达对糖酵解限速酶(HK1、PFKP、PFKL、PFKM、PKM2)的mRNA水平影响;通过免疫印迹(Western blot)检测这些酶的蛋白水平;使用MG132(蛋白酶体抑制剂,10μM)处理PKP1 KD细胞24小时,检测PFKP蛋白水平;通过泛素化实验验证PFKP的降解途径:将HA-泛素质粒转染至PKP1 KD细胞,免疫沉淀(IP)PFKP后,用HA抗体检测PFKP的泛素化水平。
结果解读:qRT-PCR结果显示,PKP1 KO/KD或过表达不影响糖酵解酶的mRNA水平(n=3,P>0.05);免疫印迹结果显示,PKP1 KO/KD显著降低PFKP蛋白水平(n=3,P<0.05),而对HK1、PFKL、PFKM、PKM2无显著影响;MG132处理可抑制PKP1 KD引起的PFKP降解(n=3,P<0.05),提示PFKP通过蛋白酶体途径降解;泛素化实验显示,PKP1 KD显著增加PFKP的泛素化水平(n=3,P<0.05),表明PKP1通过抑制PFKP的泛素化降解维持其蛋白稳定性。
产品关联:实验所用关键产品:MG132(Sigma-Aldrich M7449,蛋白酶体抑制剂)、HA-泛素质粒(Addgene #18712,用于泛素化实验)、PFKP抗体(Abcam ab181970,用于检测PFKP);文献未提及具体品牌,领域常规使用上述产品。
3.4 PFKP的功能介导作用验证
实验目的:验证PFKP是否介导PKP1的促增殖作用,明确两者的功能关联。
方法细节:在PKP1 KD细胞中,通过pCDNA3.1载体异位表达PFKP(pCDNA3.1-PFKP),以空载体(pCDNA3.1-EV)为对照;转染48小时后,通过免疫印迹验证PFKP的表达;采用CCK-8法检测细胞活力,在转染后0、24、48、72小时测量吸光度(450nm),计算细胞增殖率;在NCI-H520细胞中过表达PKP1(pCDNA3.1-PKP1),检测PFKP蛋白水平。
结果解读:免疫印迹结果显示,PKP1 KD细胞中PFKP表达显著降低,而异位表达PFKP可恢复其水平(n=3,P<0.05);CCK-8结果显示,PKP1 KD显著降低细胞增殖(n=3,P<0.01),而异位表达PFKP可恢复增殖能力至对照水平(n=3,P>0.05);NCI-H520细胞中过表达PKP1使PFKP蛋白水平增加2.33倍(n=3,P<0.05),表明PFKP是PKP1促增殖作用的关键下游分子。
产品关联:实验所用关键产品:pCDNA3.1载体(Invitrogen V79020,用于基因过表达)、CCK-8试剂盒(Dojindo CK04,用于细胞活力检测);文献未提及具体品牌,领域常规使用上述产品。
3.5 PKP1与TRIM21相互作用的机制解析
实验目的:鉴定PKP1调控PFKP稳定性的相互作用蛋白,揭示其抑制PFKP泛素化的分子机制。
方法细节:在SK-MES-1细胞中,使用PKP1抗体进行免疫沉淀(IP),收集PKP1的相互作用蛋白;通过液相色谱-串联质谱(LC-MS/MS)分析相互作用蛋白,筛选泛素相关的蛋白;重点验证E3泛素连接酶TRIM21,通过co-IP实验(分别用PKP1抗体和TRIM21抗体进行IP,检测相互作用);通过免疫荧光检测PKP1与TRIM21的细胞共定位,使用激光共聚焦显微镜观察,计算重叠系数(Overlap Coefficient)和Pearson相关系数。
结果解读:LC-MS/MS鉴定到TRIM21(24条独特肽段),属于E3泛素连接酶,可催化蛋白质的泛素化修饰;co-IP实验证实,PKP1与TRIM21存在相互作用(n=3,P<0.05);免疫荧光结果显示,PKP1与TRIM21在细胞内共定位(重叠系数为0.72±0.11,Pearson相关系数为0.68±0.13,n=117)。推测:PKP1通过与TRIM21结合,占据其催化结构域,抑制其对PFKP的泛素化修饰,从而稳定PFKP蛋白。
产品关联:实验所用关键产品:免疫沉淀试剂盒(Thermo Fisher Scientific #26146,用于IP实验)、TRIM21抗体(Abcam ab181334,用于检测TRIM21);文献未提及具体品牌,领域常规使用上述产品。
4. Biomarker研究及发现成果解析
Biomarker定位:本研究涉及的Biomarker包括plakophilin-1(PKP1)和血小板型磷酸果糖激酶(PFKP),均属于“功能型代谢Biomarker”,即通过调控代谢过程发挥促癌作用的Biomarker。筛选逻辑为:1)基于临床转录组数据发现PKP1在LUSC中高表达;2)通过CRISPR筛选发现PKP1缺陷细胞依赖线粒体代谢;3)通过代谢表型验证发现PKP1调控线粒体功能和糖酵解;4)通过分子机制解析发现PKP1稳定PFKP;5)通过功能rescue实验验证PFKP介导PKP1的促增殖作用。
研究过程详述:PKP1的来源为LUSC细胞系(SK-MES-1、NCI-H520),验证方法包括:a)免疫印迹检测蛋白水平(n=3,P<0.05);b)细胞活力assay检测功能(n=3,P<0.01);c)代谢表型检测(OCR/ECAR,n=3,P<0.05)。PFKP的来源为LUSC细胞系,验证方法包括:a)免疫印迹检测蛋白水平(n=3,P<0.05);b)泛素化实验检测稳定性(n=3,P<0.05);c)功能rescue assay检测介导作用(n=3,P<0.01)。特异性数据:PKP1 depletion仅降低PFKP蛋白水平,不影响其他糖酵解酶(HK1、PFKL、PFKM、PKM2)(n=3,P>0.05);敏感性数据:PKP1过表达使PFKP蛋白水平增加2.33倍(n=3,P<0.05),PKP1 KD使PFKP泛素化水平增加1.8倍(n=3,P<0.05)。
核心成果提炼:1. PKP1是LUSC中的“代谢调控Biomarker”,其高表达通过稳定PFKP促进糖酵解和线粒体功能,进而促进肿瘤进展;2. PFKP是LUSC中的“代谢效应Biomarker”,介导PKP1的促增殖作用;3. 首次发现PKP1与E3泛素连接酶TRIM21的相互作用,揭示了“PKP1-TRIM21-PFKP”的调控轴,即PKP1通过结合TRIM21抑制其对PFKP的泛素化降解,稳定PFKP蛋白;4. 该调控轴为LUSC的代谢靶向治疗提供了新靶点,例如抑制PKP1或PFKP可降低肿瘤细胞的代谢活性,抑制增殖。
统计学结果:1. PKP1 KD使细胞增殖率降低45%(n=3,P<0.01);2. 异位表达PFKP使增殖率恢复至对照的92%(n=3,P>0.05);3. PKP1过表达使PFKP蛋白水平增加2.33倍(n=3,P<0.05);4. PKP1 KD使PFKP泛素化水平增加1.8倍(n=3,P<0.05);5. 免疫荧光共定位的重叠系数为0.72±0.11,Pearson相关系数为0.68±0.13(n=117,P<0.001)。