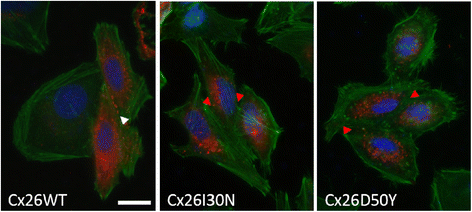
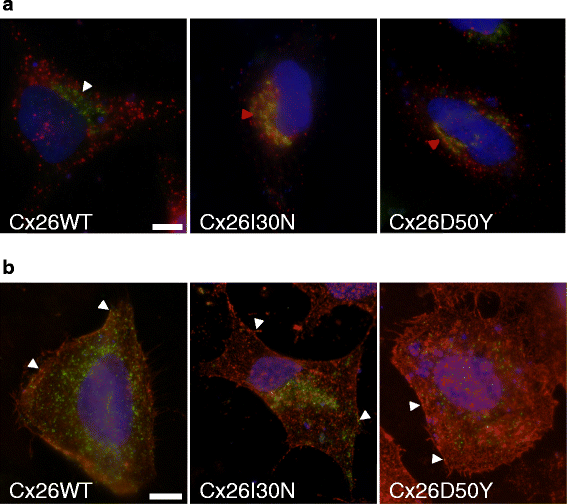
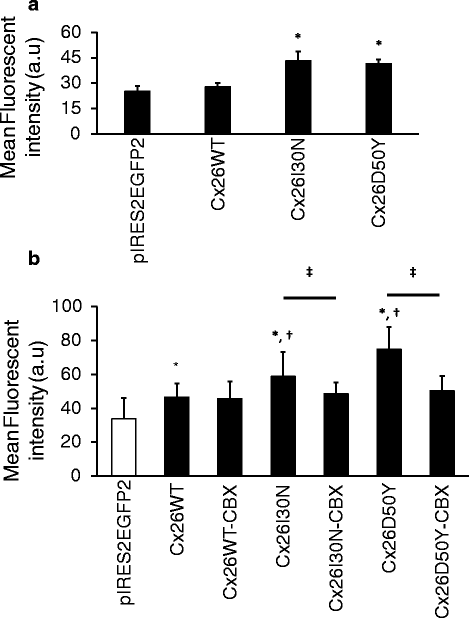
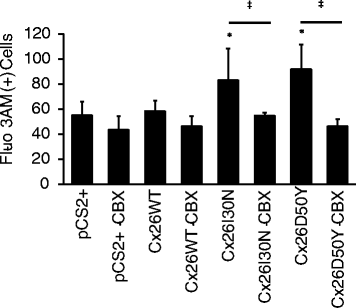
Abstract
BACKGROUND: Gap junctions facilitate exchange of small molecules between adjacent cells, serving a crucial function for the maintenance of cellular homeostasis. Mutations in connexins, the basic unit of gap junctions, are associated with several human hereditary disorders. For example, mutations in connexin26 (Cx26) cause both non-syndromic deafness and syndromic deafness associated with skin abnormalities such as keratitis-ichthyosis-deafness (KID) syndrome. These mutations can alter the formation and function of gap junction channels through different mechanisms, and in turn interfere with various cellular processes leading to distinct disorders. The KID associated Cx26 mutations were mostly shown to result in elevated hemichannel activities. However, the effects of these aberrant hemichannels on cellular processes are recently being deciphered. Here, we assessed the effect of two Cx26 mutations associated with KID syndrome, Cx26I30N and D50Y, on protein biosynthesis and channel function in N2A and HeLa cells. RESULTS: Immunostaining experiments showed that Cx26I30N and D50Y failed to form gap junction plaques at cell-cell contact sites. Further, these mutations resulted in the retention of Cx26 protein in the Golgi apparatus. Examination of hemichannel function by fluorescent dye uptake assays revealed that cells with Cx26I30N and D50Y mutations had increased dye uptake compared to Cx26WT (wild-type) containing cells, indicating abnormal hemichannel activities. Cells with mutant proteins had elevated intracellular calcium levels compared to Cx26WT transfected cells, which were abolished by a hemichannel blocker, carbenoxolone (CBX), as measured by Fluo-3 AM loading and flow cytometry. CONCLUSIONS: Here, we demonstrated that Cx26I30N and D50Y mutations resulted in the formation of aberrant hemichannels that might result in elevated intracellular calcium levels, a process which may contribute to the hyperproliferative epidermal phenotypes of KID syndrome.