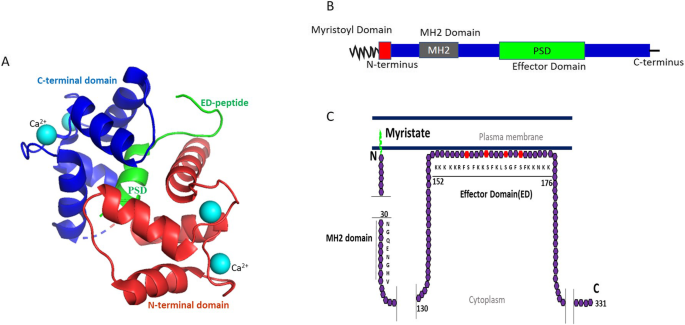
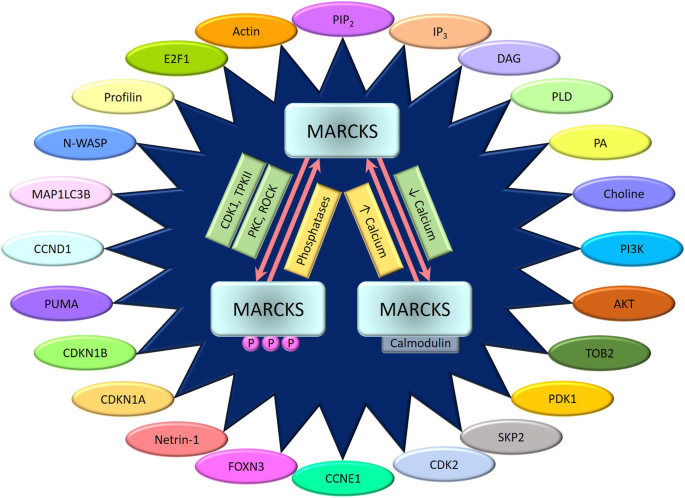
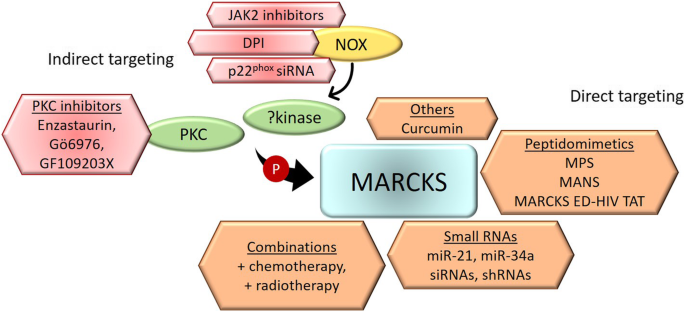
Abstract
The myristoylated alanine-rich C-kinase substrate (MARCKS) protein has been at the crossroads of multiple signaling pathways that govern several critical operations in normal and malignant cellular physiology. Functioning as a target of protein kinase C, MARCKS shuttles between the phosphorylated cytosolic form and the unphosphorylated plasma membrane-bound states whilst regulating several molecular partners including, but not limited to calmodulin, actin, phosphatidylinositol-4,5-bisphosphate, and phosphoinositide-3-kinase. As a result of these interactions, MARCKS directly or indirectly modulates a host of cellular functions, primarily including cytoskeletal reorganization, membrane trafficking, cell secretion, inflammatory response, cell migration, and mitosis. Recent evidence indicates that dysregulated expression of MARCKS is associated with the development and progression of hematological cancers. While it is understood that MARCKS impacts the overall carcinogenesis as well as plays a part in determining the disease outcome in blood cancers, we are still at an early stage of interpreting the pathophysiological roles of MARCKS in neoplastic disease. The situation is further complicated by contradictory reports regarding the role of phosphorylated versus an unphosphorylated form of MARCKS as an oncogene versus tumor suppressor in blood cancers. In this review, we will investigate the current body of knowledge and evolving concepts of the physical properties, molecular network, functional attributes, and the likely pathogenic roles of MARCKS in hematological malignancies. Key emphasis will also be laid upon understanding the novel mechanisms by which MARCKS determines the overall disease prognosis by playing a vital role in the induction of therapeutic resistance. Additionally, we will highlight the importance of MARCKS as a valuable therapeutic target in blood cancers and will discuss the potential of existing strategies available to tackle MARCKS-driven blood cancers.