1. 领域背景与文献引入
文献英文标题:Whole genome and RNA sequencing analyses for 254 Taiwanese hepatocellular carcinomas;发表期刊:Biomarker Research;影响因子:未公开;研究领域:肿瘤学-肝细胞癌多组学与精准医学
肝细胞癌(HCC)是全球第五大常见癌症、第二大癌症相关死亡原因,其发病与慢性病毒感染、酒精滥用、代谢疾病等多种因素相关,长期肝损伤和炎症可导致肝细胞坏死、再生及体细胞突变积累。领域共识:现有研究已发现HCC的常见突变基因如TP53、CTNNB1、TERT等,但多数研究基于西方或其他亚洲人群,针对台湾地区的大规模多组学研究较为缺乏。台湾地区因独特的乙肝疫苗政策、中草药使用及生活方式因素,HCC的病因谱与其他地区存在差异,其基因组特征尚未被系统揭示,同时现有HCC治疗手段疗效有限,亟需新的诊断和治疗靶点。本研究通过整合全基因组测序(WGS)和RNA测序(RNA-seq)技术,分析254例台湾HCC样本的基因组和转录组特征,旨在填补台湾HCC多组学研究的空白,为其精准诊疗提供依据。
2. 文献综述解析
作者在综述中按HCC的病因学、现有基因组研究的人群差异、非编码区与转录组研究的不足三个维度对领域研究进行分类评述。
现有研究已明确HCC的常见编码区突变基因、拷贝数变异及部分驱动通路,基于TCGA等队列的研究揭示了HCC的基因组异质性,但这些研究多集中于编码区变异,对非编码区、结构变异、可变剪接等的研究相对有限;同时,现有研究的样本多来自西方或其他亚洲人群,针对台湾地区的研究规模较小,无法反映台湾HCC的独特基因组特征;此外,现有研究对基因组变异与免疫微环境、免疫检查点基因表达的关联分析不够全面,限制了免疫治疗靶点的开发。
与现有研究相比,本研究的创新点在于首次大规模整合WGS和RNA-seq技术分析台湾HCC样本,系统揭示了台湾HCC的编码区、非编码区、结构变异、可变剪接等全维度基因组特征,明确了其与病因、预后的关联;同时,本研究深入分析了基因组变异与免疫检查点基因表达、肿瘤微环境的关联,为HCC的免疫治疗提供了新的潜在靶点;此外,本研究还揭示了台湾HCC的独特突变谱,如TERT启动子突变频率、RB1和ARID1A等基因的突变频率与TCGA队列存在差异,填补了台湾地区HCC多组学研究的空白。
3. 研究思路总结与详细解析
本研究的研究目标是系统揭示台湾HCC的基因组和转录组特征,明确其与病因、临床预后及免疫微环境的关联;核心科学问题包括台湾HCC的独特基因组变异谱是什么,这些变异如何影响患者生存和免疫微环境;技术路线遵循“样本收集→多组学测序→生物信息学分析→关联验证→结论总结”的闭环逻辑,通过整合WGS和RNA-seq数据,全面解析台湾HCC的基因组和转录组特征,并进行多维度关联分析。
3.1 样本收集与测序实验
实验目的是获取高质量的台湾HCC基因组和转录组数据,为后续分析提供基础;方法细节为收集254例手术切除的HCC肿瘤及配对癌旁组织样本,使用Qiagen试剂盒提取DNA、Macherey-Nagel试剂盒提取RNA,分别进行全基因组测序和总RNA测序,采用Illumina DRAGEN生物信息学平台进行数据处理;结果解读为成功获得254例样本的高质量WGS和RNA-seq数据,覆盖了台湾HCC的主要病因亚型(HBV相关、HCV相关、双重感染、非病毒相关、双癌相关);产品关联:文献未提及具体实验产品,领域常规使用Illumina NovaSeq系列测序平台、Qiagen DNA提取试剂盒、Macherey-Nagel RNA提取试剂盒等。
3.2 体细胞突变谱分析
实验目的是明确台湾HCC的体细胞突变特征,对比不同人群的突变差异,并分析其与病因、预后的关联;方法细节为通过生物信息学工具分析WGS数据中的体细胞突变,统计突变频率最高的基因,对比TCGA-LIHC队列的突变频率,按病因分组分析突变基因的差异,采用Kaplan-Meier法进行生存关联分析;结果解读为台湾HCC中突变频率最高的基因为TERT(47.24%,n=254),其次为TP53、CTNNB1等,与TCGA队列相比,RB1、ARID1A等基因的突变频率显著更高;不同病因组的突变基因存在差异,HCV相关HCC中FBN2、IRAK3等基因的突变频率更高(P值范围0.0024-0.0482,n=254),HBV相关HCC中ATRX突变频率更高(P=0.0367,n=254);TERT启动子突变与患者更差的总生存期相关(P=0.039,n=254);产品关联:文献未提及具体实验产品,领域常规使用Mutect2、Annovar等工具进行体细胞突变分析与注释。
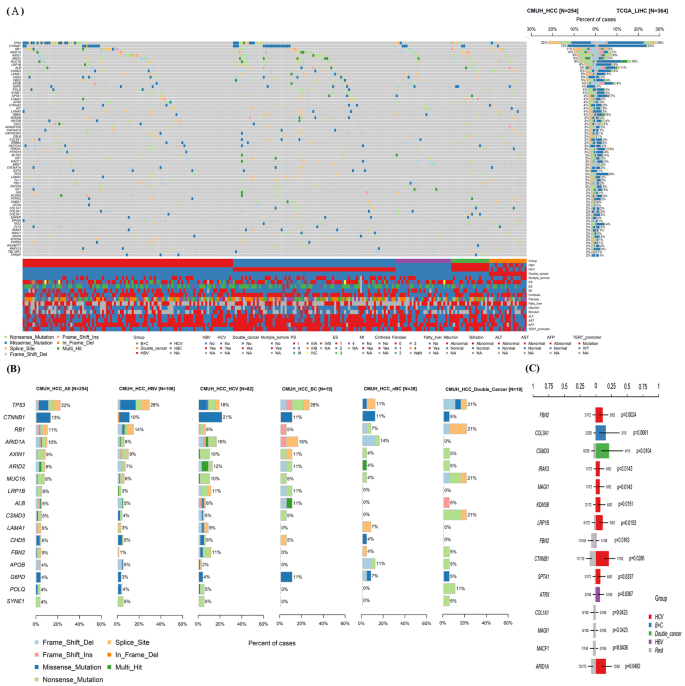
3.3 拷贝数与结构变异分析
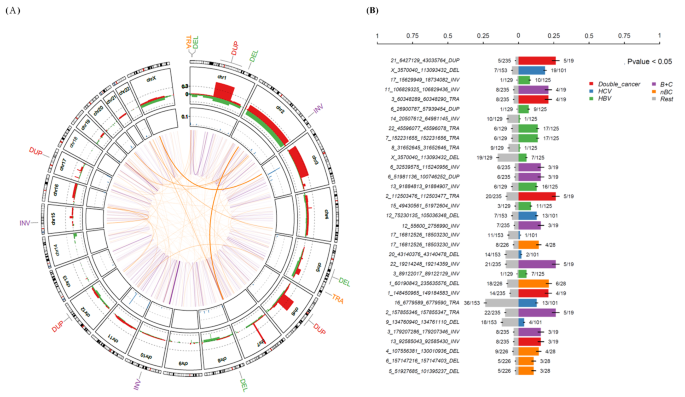
实验目的是解析台湾HCC的拷贝数变异(CNAs)和结构变异(SVs)特征,明确其与病因、预后的关联;方法细节为通过生物信息学工具分析WGS数据中的CNAs和SVs,绘制热图和Circos图展示变异分布,按病因分组分析变异频率差异,进行生存关联分析;结果解读为台湾HCC最常见的染色体臂拷贝数变异包括1q、6p等的扩增,4q、8p等的缺失,17p缺失与更好的患者生存相关(P=0.05,n=254),7q扩增与更差的生存相关(P=0.053,n=254);共鉴定出17639个体细胞SVs,其中13个SVs与患者生存显著关联,不同病因组的SVs频率存在差异;产品关联:文献未提及具体实验产品,领域常规使用GISTIC、Manta等工具进行拷贝数与结构变异分析。
3.4 非编码区与组蛋白相关基因变异分析
实验目的是探索非编码区和组蛋白相关基因在台湾HCC中的变异特征及其潜在作用;方法细节为分析114个组蛋白相关基因、74个HCC相关长链非编码RNA(lncRNAs)、36个非编码驱动基因的变异,采用Sanger测序验证HILS1基因的突变;结果解读为8.66%的台湾HCC样本存在组蛋白相关基因突变,其中HILS1的突变率最高(4例,n=254);24个HCC相关lncRNAs存在潜在驱动变异,HAGLR和LINC473的突变频率最高(各5例,n=254);非编码驱动基因中鉴定出RMRP启动子的致病性变异;产品关联:文献未提及具体实验产品,领域常规使用Sanger测序仪(如ABI 3730)进行突变验证。
3.5 转录组与可变剪接特征分析
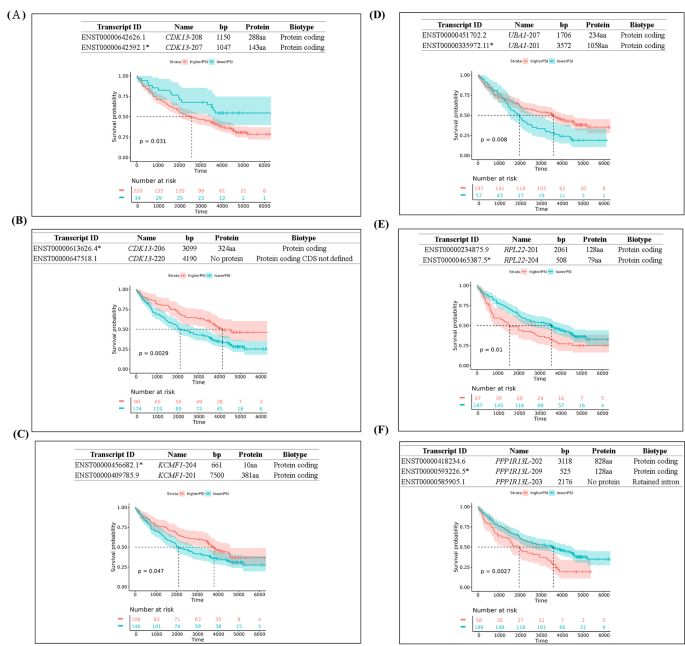
实验目的是揭示台湾HCC的转录组特征,包括差异表达基因、融合基因、可变剪接事件及其与预后的关联;方法细节为通过RNA-seq数据分析差异表达基因,鉴定融合基因,使用SUPPA2工具分析可变剪接事件,进行生存关联分析;结果解读为共鉴定出229个与患者生存相关的差异表达基因,148个新的可变剪接基因,存在融合基因的患者预后显著更好(生存曲线显示差异,n=254);不同类型的可变剪接事件对生存的影响不同,如CDK13的可变第一外显子与更好的生存相关,可变最后外显子与更差的生存相关;产品关联:文献未提及具体实验产品,领域常规使用STAR、DESeq2、SUPPA2等工具进行转录组与可变剪接分析。
3.6 基因组变异与免疫微环境关联分析
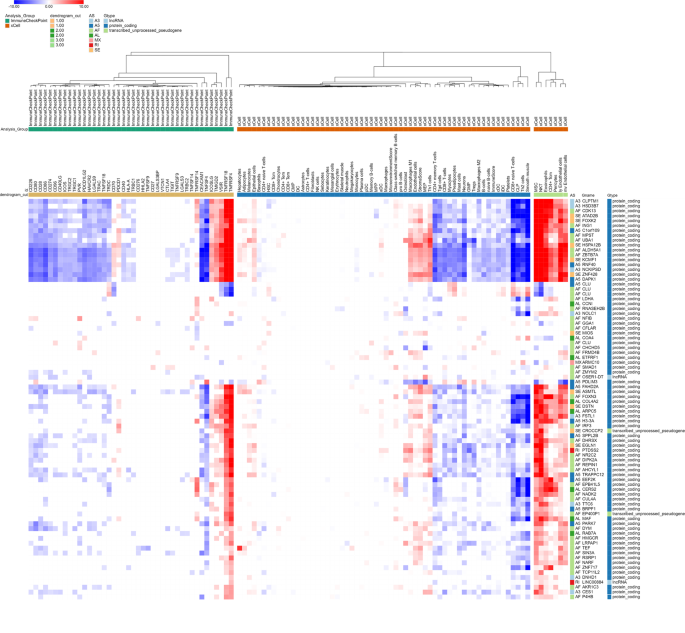
实验目的是明确台湾HCC的基因组变异与免疫检查点基因表达、肿瘤微环境的关联;方法细节为分析体细胞突变、CNAs、SVs与42个免疫检查点基因表达的关联,使用xCell工具分析基因组变异与64种免疫/基质细胞类型的关联,探索可变剪接事件与免疫微环境的关联;结果解读为ARID1A突变与CD70的高表达相关,多个基因的CNAs影响免疫检查点基因的表达(如AKT1拷贝变化影响TNFSF14表达);CTNNB1突变与上皮细胞数量减少、造血干细胞数量增加相关;生存相关的可变剪接事件与免疫细胞数量、免疫检查点基因表达显著关联,如更好生存相关的可变剪接事件与间充质干细胞、自然杀伤T细胞数量增加及TNFRSF4、PDCD1等免疫检查点基因表达上调相关;产品关联:文献未提及具体实验产品,领域常规使用xCell、CIBERSORT等工具进行肿瘤微环境分析。
4. Biomarker研究及发现成果解析
本研究通过大规模多组学分析,鉴定了一系列可用于台湾HCC诊断、预后预测及治疗指导的Biomarker,涵盖DNA水平的体细胞突变、拷贝数变异、结构变异,以及RNA水平的差异表达基因、融合基因、可变剪接基因,为HCC的精准诊疗提供了新的候选靶点。
Biomarker定位:本研究鉴定的Biomarker分为预后Biomarker和免疫治疗预测Biomarker两类,筛选逻辑为通过整合WGS和RNA-seq数据,结合生存关联分析、病因关联分析及免疫微环境关联分析,筛选出具有临床意义的基因组和转录组变异,其中部分Biomarker(如HILS1突变)经过Sanger测序验证。
研究过程详述:所有Biomarker均来自254例台湾HCC患者的肿瘤组织样本,验证方法包括多组学测序分析、生物信息学关联分析及Sanger测序验证;TERT启动子突变在台湾HCC中的突变频率为47.24%(n=254),在HCV相关HCC中的突变频率显著高于HBV相关HCC(P=5.024e-06,n=254),且与患者更差的总生存期相关(P=0.039,n=254);17p缺失与患者更好的生存相关(P=0.05,n=254);存在融合基因的患者预后显著更好(生存曲线显示差异,n=254);229个差异表达基因和148个可变剪接基因与患者生存显著关联;ARID1A突变与CD70高表达相关,可作为免疫检查点抑制剂治疗的潜在预测Biomarker。
核心成果提炼:TERT启动子突变可作为台湾HCC的预后不良Biomarker,其在HCV相关HCC中的高特异性可为病因诊断提供参考;17p缺失、存在融合基因可作为台湾HCC的预后良好Biomarker;ARID1A突变、AKT1拷贝变化等基因组变异可作为免疫检查点抑制剂治疗的潜在预测Biomarker;首次揭示了台湾HCC的独特可变剪接特征及其与免疫微环境的关联,为HCC的精准治疗提供了新的靶点;此外,组蛋白相关基因(HILS1)、非编码RNA(HAGLR、LINC473)的突变可作为HCC发病机制研究的潜在Biomarker,为病因学研究提供新方向。本研究的Biomarker覆盖了HCC诊疗的多个环节,具有较高的临床转化潜力。