Abstract
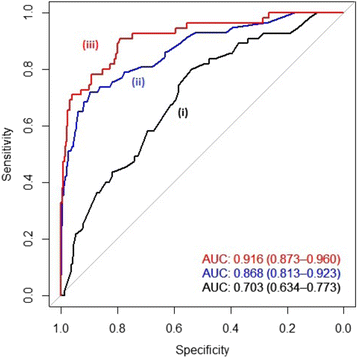
BACKGROUND: Colorectal cancer has significant impact on individuals and healthcare systems. Many genes have been identified to influence its pathogenesis. However, the genetic basis of mucinous tumor histology, an aggressive subtype of colorectal cancer, is currently not well-known. This study aimed to identify common and rare genetic variations that are associated with the mucinous tumor phenotype. METHODS: Genome-wide single nucleotide polymorphism (SNP) data was investigated in a colorectal cancer patient cohort (n = 505). Association analyses were performed for 729,373 common SNPs and 275,645 rare SNPs. Common SNP association analysis was performed using univariable and multivariable logistic regression under different genetic models. Rare-variant association analysis was performed using a multi-marker test. RESULTS: No associations reached the traditional genome-wide significance. However, promising genetic associations were identified. The identified common SNPs significantly improved the discriminatory accuracy of the model for mucinous tumor phenotype. Specifically, the area under the receiver operating characteristic curve increased from 0.703 (95% CI: 0.634-0.773) to 0.916 (95% CI: 0.873-0.960) when considering the most significant SNPs. Additionally, the rare variant analysis identified a number of genetic regions that potentially contain causal rare variants associated with the mucinous tumor phenotype. CONCLUSIONS: This is the first study applying both common and rare variant analyses to identify genetic associations with mucinous tumor phenotype using a genome-wide genotype data. Our results suggested novel associations with mucinous tumors. Once confirmed, these results will not only help us understand the biological basis of mucinous histology, but may also help develop targeted treatment options for mucinous tumors.