1. 领域背景与文献
文献英文标题:Catalogue of stable and variable interindividual DNA methylation sites in human whole blood;发表期刊:Genome Biology;影响因子:未公开;研究领域:表观遗传学(DNA甲基化个体间变异)
表观遗传学是解析遗传与环境交互作用、解释复杂表型差异的核心领域,其中DNA甲基化是最具代表性的表观遗传修饰之一。目前人类个体间遗传变异已被系统表征,但表观遗传变异的全基因组详细目录仍不完善。同卵双胞胎在遗传背景完全一致的情况下,仍存在疾病易感性、表型特征等方面的差异,这类现象无法仅用遗传序列变异和环境暴露解释,提示表观遗传变异可能是关键驱动因素。过往研究已在多种组织中鉴定到可变甲基化区域,但存在技术局限:如早期研究仅针对人类主要组织相容性复合体(MHC)等特定基因组区域,或使用覆盖度较低的450K甲基化芯片(仅约45万个CpG位点),无法全面捕捉远端调控元件的甲基化变异;部分全基因组研究样本量较小,缺乏人群代表性。本研究针对上述空白,采用覆盖85万个CpG位点的Illumina EPIC芯片,在英国代表性人群队列(3642例全血样本)中系统鉴定稳定与可变甲基化位点,构建了全基因组范围的全血DNA甲基化变异目录,为表观基因组关联研究(EWAS)的结果解读提供关键资源,同时解析了甲基化变异的调控机制与功能意义。
2. 文献综述解析
作者对领域内现有研究的分类维度主要包括研究的组织类型、技术平台(芯片/测序)、研究范围(特定区域/全基因组)及核心研究方向(变异鉴定/机制解析)。现有研究的关键结论可整合为:DNA甲基化个体间变异与表型差异密切相关,同卵双胞胎的疾病不一致性已被证实与表观遗传差异直接关联;不同组织中的可变甲基化区域具有显著组织特异性,且多富集在调控元件区域;甲基化数量性状位点(mQTLs)可作为遗传变异与表观遗传变异的桥梁,解析遗传对甲基化的调控作用。技术方法方面,全基因组亚硫酸氢盐测序可实现单碱基分辨率检测,但因成本高昂导致样本量普遍较小;450K甲基化芯片虽降低了研究成本,但对启动子以外的远端调控元件覆盖不足;部分研究针对特定疾病队列,虽能揭示疾病相关甲基化变异,但缺乏普通人群的代表性数据。现有研究的核心局限性在于:缺乏大样本量、人群代表性的全基因组全血甲基化变异目录;对可变甲基化位点的功能调控机制(如与转录因子结合、3D基因组结构的关联)解析不够深入;对表观等位基因(epialleles)的系统鉴定及功能验证仍较匮乏。
本研究的创新价值通过与现有研究的对比凸显:首次采用覆盖度更高的EPIC芯片在3642例大样本代表性人群中系统鉴定全血的稳定与可变甲基化位点,填补了全基因组范围全血甲基化变异目录的空白;深入解析了可变甲基化位点与转录因子结合、mQTLs及3D基因组结构的关联,揭示了甲基化变异的多层调控机制;系统鉴定了784个潜在表观等位基因,并明确其与基因表达的调控关系,为表观遗传变异驱动表型差异提供了直接证据。
3. 研究思路总结与详细解析
本研究的整体框架为:以英国代表性人群全血样本为研究对象,通过大样本发现-验证队列设计,系统鉴定全基因组范围的稳定与可变甲基化位点;从基因组注释、转录因子调控、遗传关联、3D基因组结构等多维度解析甲基化变异的调控机制;进一步鉴定潜在表观等位基因并验证其对基因表达的调控作用,形成“变异鉴定-机制解析-功能验证”的完整研究闭环。核心科学问题聚焦于:人类全血中DNA甲基化个体间变异的基因组特征是什么?遗传、转录因子、3D基因组结构如何调控这些变异?甲基化变异是否及如何影响基因表达?
3.1 样本处理与数据预处理
实验目的是获得高质量、无偏倚的DNA甲基化数据集,确保后续变异鉴定的可靠性。方法细节:选取Understanding Society队列的全血样本,分为发现集(1171例)和验证集(2471例),采用Illumina EPIC芯片检测DNA甲基化;数据预处理流程包括:去除SNP探针、交叉杂交探针、性染色体探针,使用dasen法校正I型和II型探针的偏倚,通过甲基化性别分类器去除报告性别与生物学性别不一致的样本,利用MethylToSNP包去除与SNP位点混淆的CpG位点,最终保留747302个CpG位点用于后续分析。结果解读:经过严格质控与预处理,获得了覆盖全基因组的大样本甲基化数据集,队列的人群代表性确保了研究结果可推广至英国普通人群。产品关联:文献未提及具体实验产品,领域常规使用Illumina Infinium MethylationEPIC BeadChip芯片、R语言的bigmelon、minfi等数据处理包完成样本检测与数据预处理。
3.2 稳定与可变甲基化位点的鉴定
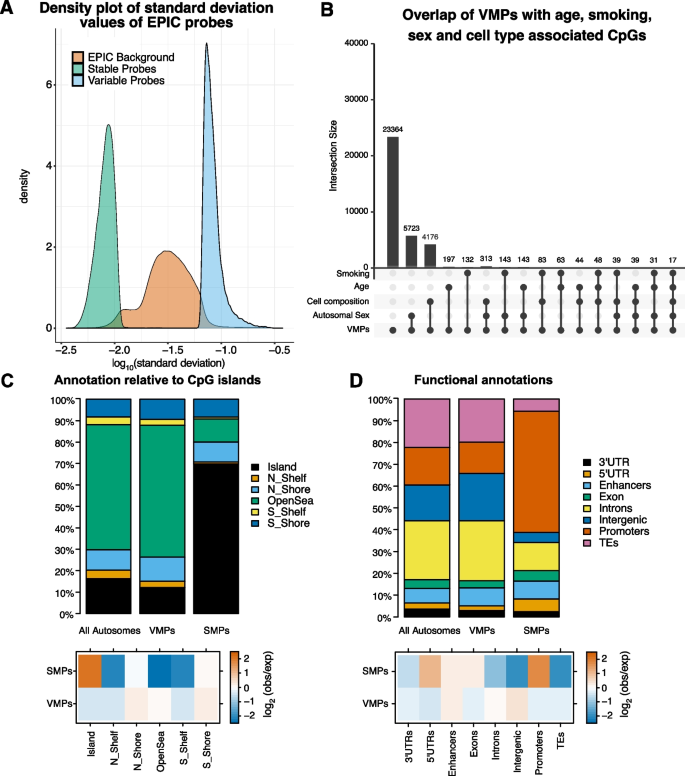
实验目的是在全基因组范围精准鉴定个体间甲基化水平稳定(SMPs)与可变(VMPs)的CpG位点。方法细节:采用降采样法控制样本偏倚,对发现集随机去除10%样本后,计算每个CpG位点甲基化值的标准差(SD),将前10%高SD位点定义为VMPs,后10%低SD位点定义为SMPs;重复该过程10次,仅保留所有重复中均出现的位点;在验证集中重复相同分析流程,最终仅保留在发现集和验证集均鉴定到的位点。结果解读:共鉴定得到34972个VMPs和41216个SMPs;VMPs主要呈现中等甲基化水平,而SMPs以高或低甲基化水平为主;约50%的VMPs为EPIC芯片特有,无法通过450K芯片检测;细胞类型组成仅解释不到15%的VMPs变异,说明甲基化变异主要由遗传、环境或随机表观遗传过程驱动。
产品关联:文献未提及具体实验产品,领域常规使用R语言的统计分析包完成标准差计算与降采样分析。
3.3 VMPs/SMPs的基因组注释与功能富集
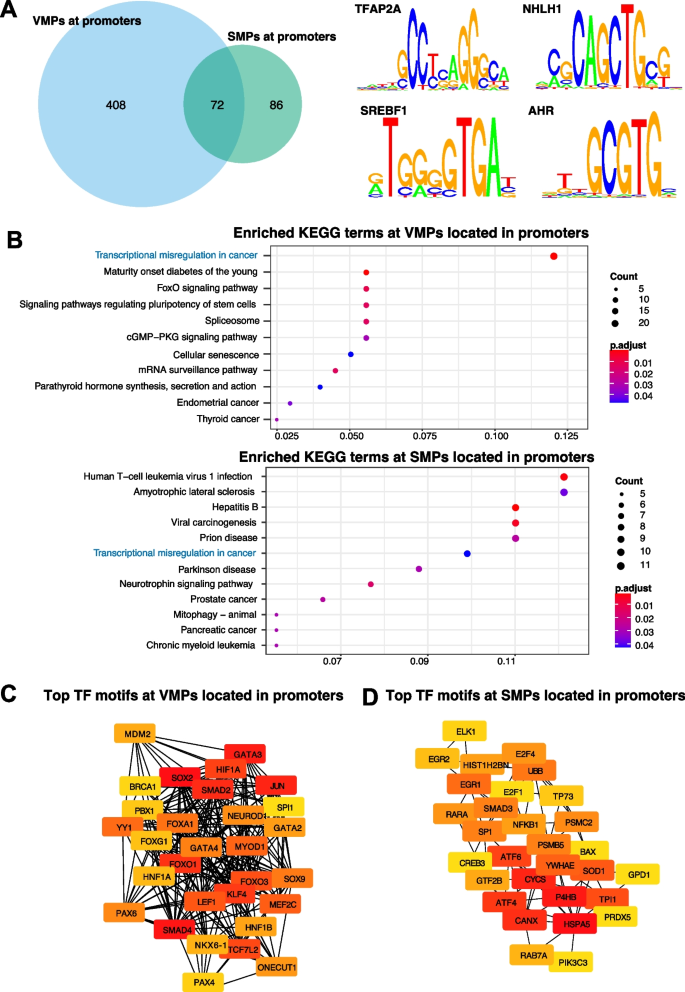
实验目的是解析VMPs与SMPs的基因组分布特征及潜在生物学功能。方法细节:利用Minfi包将CpG位点注释到CpG岛、启动子、增强子、基因区域等基因组元件;采用PWMEnrich包进行转录因子结合基序富集分析,以SMPs为背景分析VMPs的特有富集基序,反之亦然;通过GO和KEGG通路分析富集基序的功能关联;同时分析VMPs/SMPs与年龄、吸烟、性别相关CpG位点的重叠,以及与印记区域的关联。结果解读:VMPs显著富集在CpG岸、增强子、基因间区,而SMPs主要富集在CpG岛、启动子区域及管家基因;VMPs富集的转录因子基序以TFAP2A、NHLH1为主,SMPs富集的基序以SREBF1、AHR为主,且这些转录因子多为甲基化敏感型,提示甲基化变异可通过影响转录因子结合调控基因表达;VMPs与年龄、吸烟、性别相关CpG的重叠率高于SMPs,但仍有大部分VMPs不与这些已知表型相关CpG重叠,提示存在未被发现的调控因素;VMPs与印记区域显著重叠(242个位点),而SMPs仅1个位点与印记区域重叠,提示可变甲基化可能参与等位基因特异性调控。
产品关联:文献未提及具体实验产品,领域常规使用R语言的PWMEnrich、clusterProfiler等包完成功能富集分析。
3.4 VMPs/SMPs与mQTLs及3D基因组结构的关联
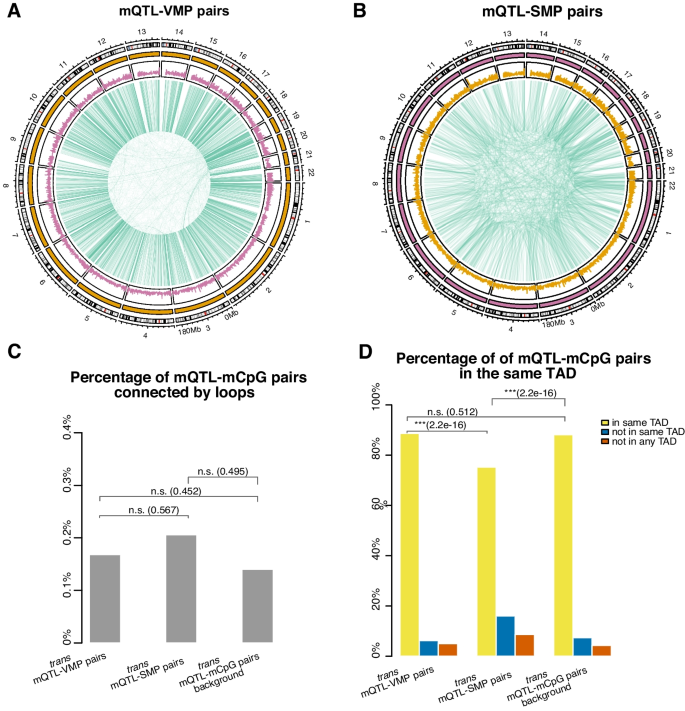
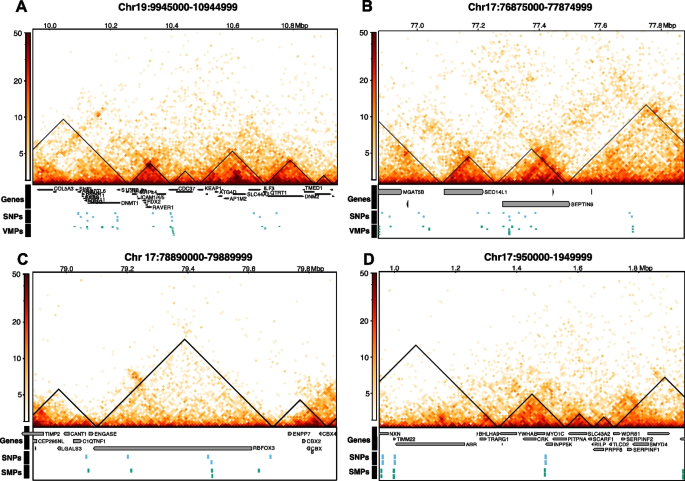
实验目的是解析遗传变异对DNA甲基化个体间变异的调控作用,以及3D基因组结构在其中的介导作用。方法细节:将VMPs/SMPs与已发表的全血mQTLs数据关联,定义顺式(cis)mQTL为SNP与CpG位点距离≤500bp的对,反式(trans)mQTL为距离>500bp的对;利用全血Hi-C数据,分析trans mQTL对是否位于同一拓扑关联域(TAD)或通过染色质环连接。结果解读:44.9%的VMPs与mQTLs关联,其中21%为cis mQTL,79%为trans mQTL;仅3.27%的SMPs与mQTLs关联,提示可变甲基化位点受遗传调控的比例远高于稳定位点;大部分trans mQTL-VMP对位于同一TAD,说明3D基因组结构可介导遗传变异对远端甲基化位点的调控;mQTL-VMP关联的SNP富集在细胞形态发生、小GTP酶信号转导等生物学通路,进一步支持遗传对甲基化的功能调控。
产品关联:文献未提及具体实验产品,领域常规使用Juicer、HiCExplorer等工具完成3D基因组结构分析。
3.5 潜在表观等位基因的鉴定与功能分析
实验目的是鉴定全血中的潜在表观等位基因,并解析其对基因表达的调控作用。方法细节:从VMPs中筛选平均甲基化水平在0.4-0.6之间的位点,采用Hartigan’s dip test检测甲基化分布的双峰性,将符合条件的位点定义为潜在表观等位基因;分析表观等位基因与mQTLs的关联、组织特异性(与脑、骨骼肌样本的重叠率);利用SHIP-Trend队列的基因表达芯片数据、CD14+单核细胞的RNA-seq数据,分别分析甲基化变异与基因表达变异的相关性。结果解读:共鉴定得到784个潜在表观等位基因,其中58%为EPIC芯片特有;81%的表观等位基因与mQTLs关联,且以trans mQTL为主;表观等位基因在不同组织间的重叠率极低,提示其具有显著组织特异性;在非匹配人群队列中,位于5’UTR的表观等位基因的甲基化变异与靶基因表达变异呈正相关;在匹配的CD14+单核细胞队列中,5’UTR区域的表观等位基因甲基化与基因表达呈负相关,启动子区域的表观等位基因(如TACSTD2、Xist启动子)显示出强负相关,直接证明甲基化变异对基因表达的调控作用。
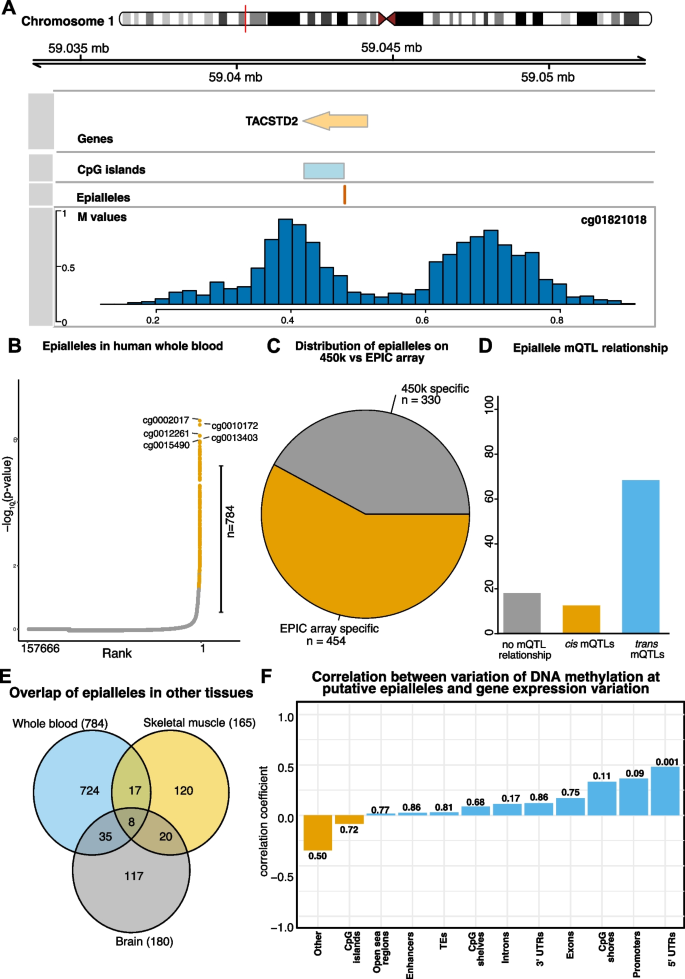
产品关联:文献未提及具体实验产品,领域常规使用R语言的diptest包完成双峰分布检测,通过Pearson相关分析甲基化与基因表达的关联。
4. Biomarker研究及发现成果
本研究中涉及的Biomarker主要包括全血DNA甲基化可变位点(VMPs)和潜在表观等位基因两类。VMPs的定位为人类全血中个体间甲基化水平存在显著变异的CpG位点,筛选逻辑为:在发现集与验证集的大样本队列中,通过重复降采样分析均被鉴定为高标准差的位点;潜在表观等位基因的定位为VMPs中具有双峰甲基化分布的位点,筛选逻辑为:平均甲基化水平在0.4-0.6之间,且经Hartigan’s dip test检测显示显著双峰分布(p<0.05)。
研究过程方面,两类Biomarker均来自人类全血样本,采用Illumina EPIC芯片进行甲基化检测;VMPs通过发现集与验证集的重复鉴定确保可靠性,潜在表观等位基因通过组织特异性分析、mQTL关联分析进一步验证其生物学意义;特异性上,VMPs显著富集在调控元件区域,与管家基因无关联,潜在表观等位基因具有显著组织特异性;敏感性方面,大样本量(3642例)确保了Biomarker鉴定的统计学效力。
核心成果提炼:VMPs目录为EWAS研究提供了关键参考,仅针对可变位点进行分析可显著提高统计效力,同时为结果解读提供背景依据;潜在表观等位基因中部分位点的甲基化变异可直接调控基因表达,为表型差异的表观遗传机制提供了直接证据;约50%的VMPs受遗传变异调控,15%的变异由细胞类型组成驱动,约四分之一的VMPs与年龄、性别、吸烟等已知表型相关因素关联;784个潜在表观等位基因中,约19%未与任何SNP关联,提示其可能由随机表观遗传过程或环境暴露驱动。具体数据包括:VMPs数量为34972个(n=3642),SMPs数量为41216个(n=3642);潜在表观等位基因数量为784个(n=3642,Hartigan’s dip test p<0.05);44.9%的VMPs与mQTLs关联(n=3642,关联具有统计学显著性);位于5’UTR的表观等位基因甲基化变异与基因表达变异的Pearson相关系数在非匹配队列中呈正相关,在匹配队列中呈负相关(p<0.05)。