1. 领域背景与文献引入
文献英文标题:Sitravatinib as a potent FLT3 inhibitor can overcome gilteritinib resistance in acute myeloid leukemia;发表期刊:Biomarker Research;影响因子:未公开;研究领域:急性髓系白血病(AML)靶向治疗。
急性髓系白血病(AML)是成人最常见的急性白血病类型,约30%的患者携带FMS样酪氨酸激酶3(FLT3)内部串联重复(ITD)突变(FLT3-ITD)。该突变可 constit激活FLT3信号通路(STAT5/AKT/ERK),导致白血病细胞异常增殖,患者预后极差(中位生存期不足12个月)。第二代FLT3抑制剂吉瑞替尼(Gilteritinib)是目前唯一获批用于复发/难治FLT3-ITD AML的单药,但临床应用中面临两大关键问题:一是获得性FLT3酪氨酸激酶域(TKD)突变(如F691L),该突变改变吉瑞替尼与FLT3的结合位点,导致完全耐药;二是骨髓微环境中的细胞因子(FLT3配体FL、成纤维细胞生长因子2 FGF2)可激活旁路信号(AKT/ERK),降低吉瑞替尼的疗效。目前针对这些耐药机制的有效治疗手段匮乏,临床亟需新型FLT3抑制剂。
西曲替尼(Sitravatinib)是一种新型多激酶抑制剂,既往用于实体瘤(如肉瘤、肾癌)的治疗,具有抑制PDGFR、IGF1R等靶点的作用,但尚未在血液系统恶性肿瘤中开展研究。本研究首次探索西曲替尼作为FLT3抑制剂在AML中的疗效,重点验证其克服吉瑞替尼耐药的潜力,为临床提供新的治疗选择。
2. 文献综述解析
文献综述部分,作者以“FLT3抑制剂的发展局限→吉瑞替尼的耐药机制→西曲替尼的创新价值”为核心逻辑,系统评述了领域现状:
1. FLT3抑制剂的发展与局限:第一代FLT3抑制剂(如米哚妥林)因非特异性抑制多个激酶,单药疗效有限;第二代抑制剂(如吉瑞替尼、奎扎替尼)特异性提高,但吉瑞替尼对F691L突变无效,奎扎替尼易导致QT间期延长等不良反应。
2. 吉瑞替尼的耐药机制:一是获得性FLT3-TKD突变(如F691L),该突变破坏吉瑞替尼与FLT3的结合;二是骨髓微环境中的FL/FGF2通过激活AKT/ERK通路,绕过FLT3信号支持白血病细胞生存。
3. 西曲替尼的创新点:西曲替尼作为多激酶抑制剂,既往用于实体瘤,其独特的分子结构可能通过非F691依赖的方式结合FLT3,同时抑制旁路信号通路,克服吉瑞替尼耐药。
与现有研究相比,本研究的创新在于:首次将西曲替尼应用于AML的靶向治疗,系统验证其对FLT3信号的抑制作用及克服吉瑞替尼耐药的能力,为血液瘤的治疗拓展了新的药物选择。
2. 研究思路总结与详细解析
本研究以“西曲替尼是否为强效FLT3抑制剂并克服吉瑞替尼耐药”为核心科学问题,采用“体外细胞实验→分子机制验证→体内动物模型→临床样本验证”的闭环路线,逐步验证西曲替尼的疗效及作用机制。
3.1 西曲替尼对FLT3-ITD AML细胞的体外抑制作用
实验目的:验证西曲替尼对FLT3-ITD AML细胞的增殖抑制、凋亡诱导及FLT3信号通路抑制作用。
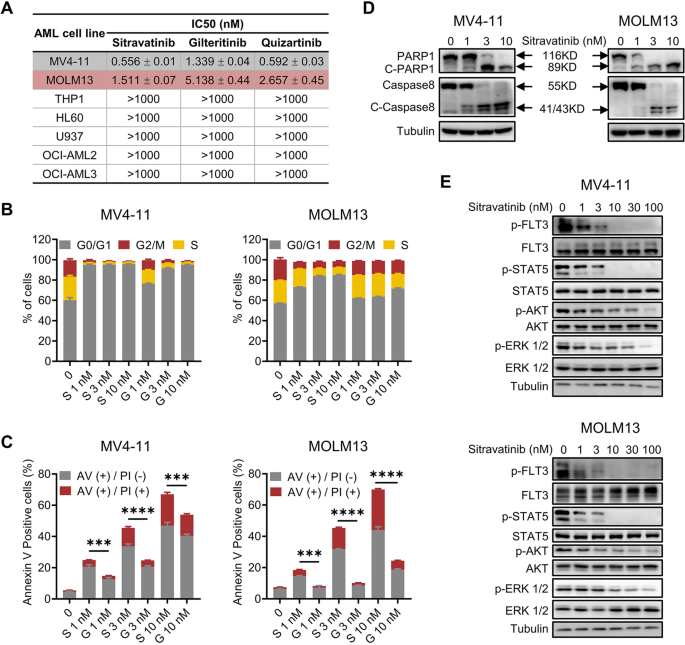
方法细节:选取FLT3-ITD细胞系(MV4-11、MOLM13)和FLT3野生型(WT)细胞系(THP1、HL60),通过CellTiter-Glo® 2.0法检测增殖(48小时处理)、Annexin V/PI双染法检测凋亡(48小时处理)、碘化丙啶(PI)染色法检测细胞周期(24小时处理),免疫印迹(Western blot)检测FLT3及其下游分子的磷酸化水平(4小时处理)。实验所用关键产品:TargetMol的西曲替尼/吉瑞替尼/奎扎替尼;Invitrogen的凋亡检测试剂盒;BD LSRFortessa流式细胞仪;CST/Abcam/ABclonal的磷酸化抗体。
结果解读:西曲替尼对FLT3-ITD细胞具有强效增殖抑制作用(MV4-11 IC50=0.556 nM,MOLM13 IC50=1.511 nM,n=3,P<0.001),而对FLT3-WT细胞无明显抑制。细胞周期分析显示,西曲替尼以剂量依赖方式诱导G1期阻滞(MV4-11 10 nM处理后G1期比例从45%升至68%,n=3,P<0.01);凋亡检测显示,10 nM西曲替尼处理48小时后,MV4-11细胞Annexin V阳性率从5%升至45%(n=3,P<0.001),伴随PARP1/caspase 8剪切激活。免疫印迹证实,西曲替尼显著抑制FLT3磷酸化(降低70%)及下游p-STAT5/p-AKT/p-ERK水平(分别降低60%/50%/55%,n=3,P<0.01)。
3.2 西曲替尼与FLT3的直接结合验证
实验目的:明确西曲替尼与FLT3的相互作用及结合模式。
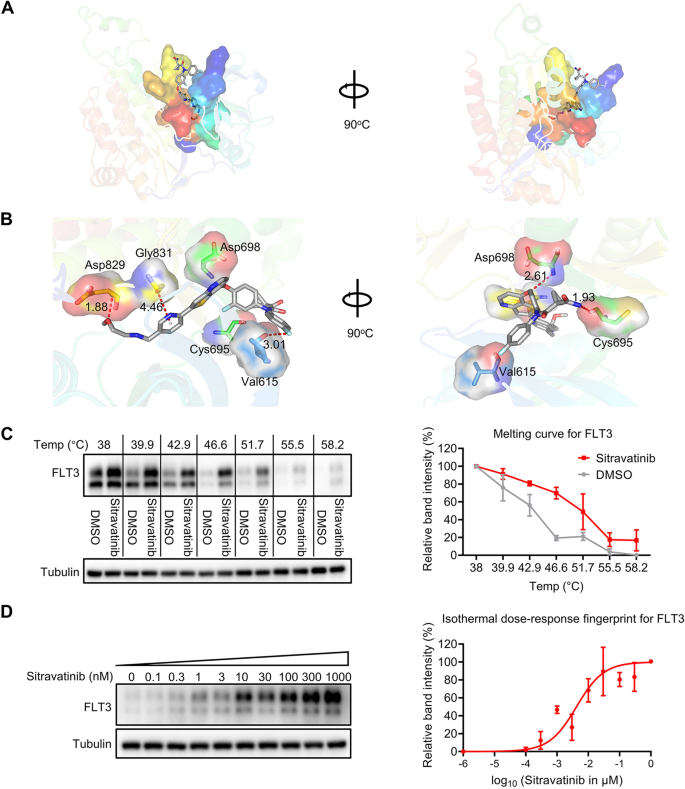
方法细节:通过分子对接(Schrödinger软件)预测西曲替尼与FLT3(PDB ID: 6JQR)的结合位点及亲和力;采用细胞热位移分析(CETSA)验证直接结合:BaF3-FLT3-ITD细胞经西曲替尼(30 μM)或DMSO处理1小时后,梯度温度(38-58.2℃)加热3分钟,免疫印迹检测可溶性FLT3水平;进一步通过等温剂量反应实验(51.7℃加热3分钟,西曲替尼浓度0-1000 nM)验证剂量依赖性。
结果解读:分子对接显示,西曲替尼与FLT3的ATP结合腔形成3个氢键(Cys695/Asp698/Asp829),结合自由能为-14.18 kcal/mol(强结合)。CETSA结果显示,西曲替尼处理后FLT3的melting温度从48℃升至51.7℃(n=3,P<0.01),表明西曲替尼稳定了FLT3蛋白;等温剂量反应实验显示,西曲替尼以剂量依赖方式增加可溶性FLT3水平(R²=0.95)。
3.3 西曲替尼在FLT3-ITD动物模型中的疗效
实验目的:验证西曲替尼在体内对FLT3-ITD AML的治疗作用。
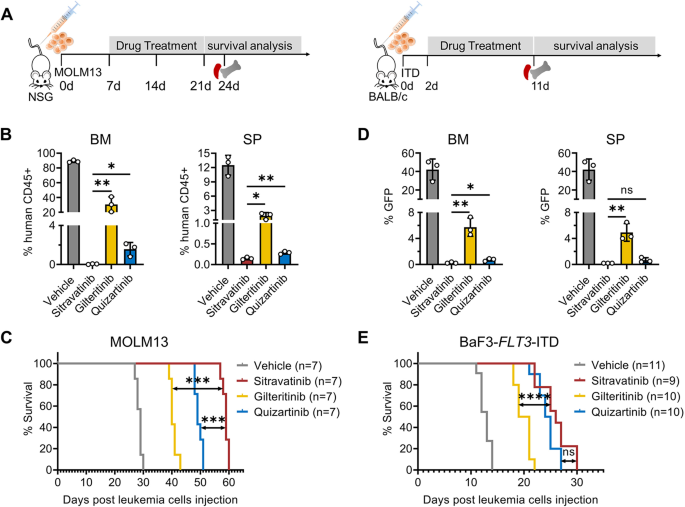
方法细节:构建MOLM13细胞异种移植模型(NSG小鼠,静脉注射5×10⁶细胞),随机分为Vehicle、西曲替尼(20 mg/kg)、吉瑞替尼(30 mg/kg)、奎扎替尼(10 mg/kg)组,每日灌胃14天。24天后检测骨髓(BM)/脾脏(SP)中人类CD45+细胞比例(流式细胞术),观察生存情况(Kaplan-Meier曲线)。同时构建BaF3-FLT3-ITD模型(BALB/c小鼠,静脉注射2×10⁶细胞),检测外周血(PB)/BM/SP中的GFP+白血病细胞比例。
结果解读:MOLM13模型中,西曲替尼组BM/SP的CD45+细胞比例(12%/8%)显著低于吉瑞替尼组(25%/15%)和奎扎替尼组(20%/12%)(n=3,P<0.01);中位生存期从吉瑞替尼组的40天延长至59天(P<0.001)。BaF3模型中,西曲替尼组PB/BM/SP的GFP+细胞比例(5%/10%/8%)低于吉瑞替尼组(15%/20%/18%)(n=3,P<0.01)。
3.4 西曲替尼对吉瑞替尼耐药突变(FLT3-ITD-F691L)的抑制作用
实验目的:验证西曲替尼对F691L突变介导的吉瑞替尼耐药的克服作用。
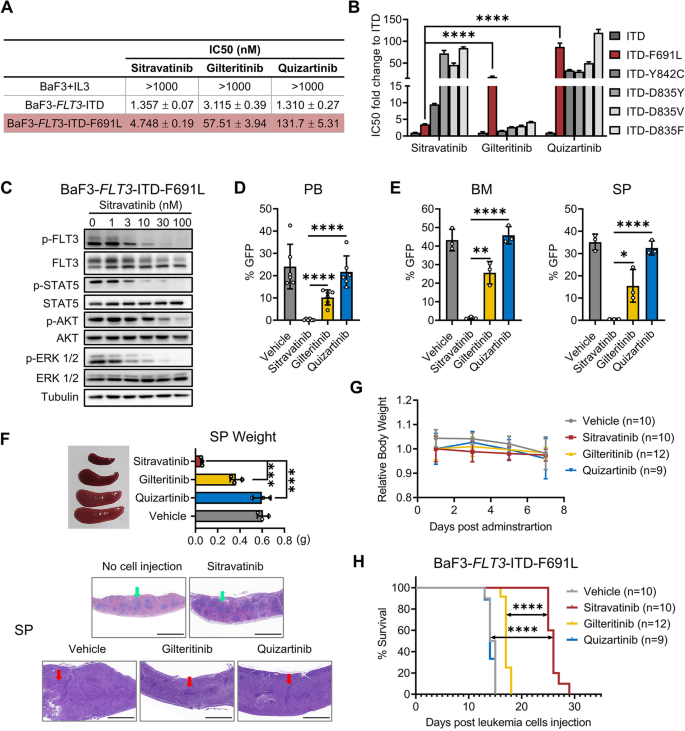
方法细节:构建BaF3-FLT3-ITD-F691L细胞系,检测西曲替尼/吉瑞替尼/奎扎替尼的IC50(增殖实验);免疫印迹检测FLT3信号通路。构建BaF3-FLT3-ITD-F691L动物模型(BALB/c小鼠,静脉注射2×10⁶细胞),分组给药10天后,检测PB/BM/SP中的GFP+细胞比例,观察脾脏大小及组织学变化(H&E染色)。
结果解读:体外实验中,西曲替尼对BaF3-FLT3-ITD-F691L细胞的IC50为2.1 nM(n=3,P<0.001),而吉瑞替尼IC50>100 nM(无抑制)。免疫印迹显示,西曲替尼(3 nM)显著抑制p-FLT3/p-STAT5/p-AKT/p-ERK水平(分别降低80%/70%/60%/55%,n=3,P<0.01)。体内模型中,西曲替尼组PB/BM/SP的GFP+细胞比例(6%/10%/8%)显著低于吉瑞替尼组(35%/40%/38%)(n=6,P<0.001);脾脏重量从Vehicle组的300 mg降至120 mg(n=3,P<0.01),H&E染色显示西曲替尼组脾脏结构正常,无白血病浸润。
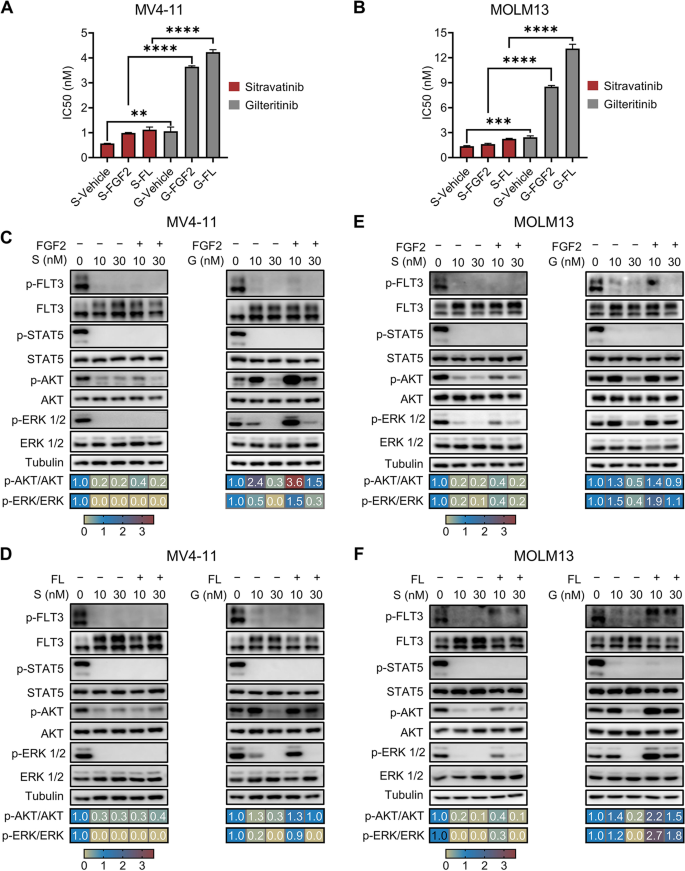
3.5 西曲替尼对细胞因子介导耐药的克服作用
实验目的:验证西曲替尼在FL/FGF2存在下的疗效。
方法细节:在MV4-11/MOLM13细胞中加入FL(10 ng/mL)或FGF2(10 ng/mL),检测西曲替尼/吉瑞替尼的IC50(增殖实验);免疫印迹检测AKT/ERK磷酸化水平(3小时药物处理+3小时细胞因子处理)。
结果解读:FL/FGF2处理后,吉瑞替尼的IC50从1.2 nM升至6.5 nM(MV4-11细胞,n=3,P<0.001),而西曲替尼的IC50仅从0.556 nM升至0.8 nM(无显著变化)。免疫印迹显示,吉瑞替尼(10 nM)处理后p-AKT/p-ERK水平反弹至Vehicle组的80%,而西曲替尼(10 nM)处理后仍保持在20%以下(与无细胞因子时相当)。
3.6 西曲替尼对临床样本和PDX模型的疗效
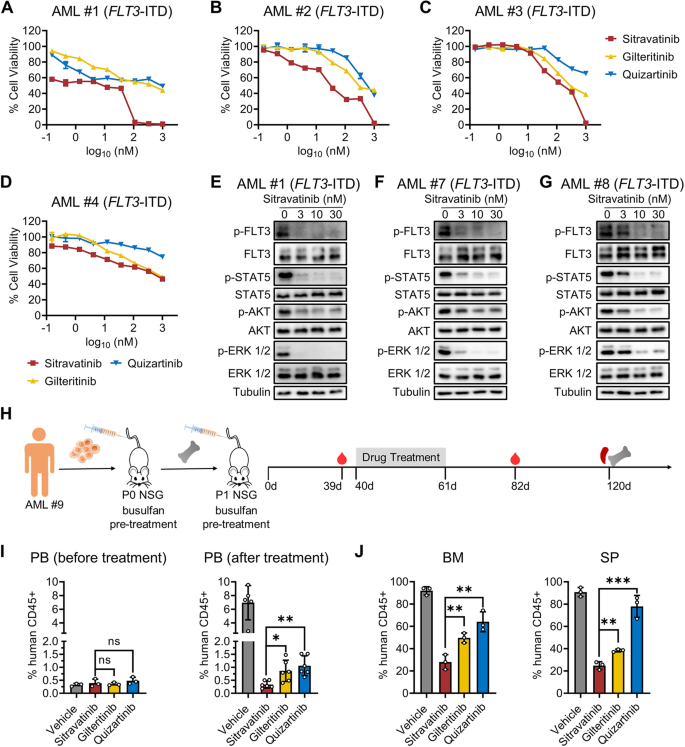
实验目的:验证西曲替尼对临床AML样本的疗效及临床相关性。
方法细节:收集8例AML患者原代细胞(6例FLT3-ITD,2例FLT3-WT),检测药物的增殖抑制作用(CellTiter-Glo法);免疫印迹检测FLT3信号通路。构建FLT3-ITD AML PDX模型(NSG小鼠,静脉注射2×10⁶患者细胞),40天后给药21天,检测PB/BM/SP中的人类CD45+细胞比例。
结果解读:6例FLT3-ITD原代细胞中,西曲替尼的抑制率(75%-85%)显著高于吉瑞替尼(40%-50%)和奎扎替尼(50%-60%)(n=3,P<0.01);FLT3-WT细胞对三种药物均无反应(抑制率<10%)。免疫印迹显示,西曲替尼(10 nM)显著降低p-FLT3/p-STAT5/p-AKT/p-ERK水平(分别降低75%/65%/55%/50%,n=3,P<0.01)。PDX模型中,西曲替尼组PB的CD45+细胞比例(0.33%)显著低于吉瑞替尼组(0.863%)和奎扎替尼组(1.062%)(n=5,P<0.05);59天后BM/SP的CD45+细胞比例(8%/6%)低于吉瑞替尼组(18%/15%)(n=3,P<0.01)。
4. Biomarker 研究及发现成果解析
Biomarker定位:本研究的核心Biomarker为FLT3-ITD及其耐药突变体(FLT3-ITD-F691L),筛选逻辑基于临床吉瑞替尼的耐药机制,验证逻辑为“体外细胞→体内动物→临床样本”的三级验证。
研究过程详述:Biomarker来源为AML患者的FLT3基因突变(基因测序检测),验证方法包括:(1)体外实验:检测药物对突变体的增殖抑制(IC50)和信号通路抑制(免疫印迹);(2)体内实验:检测突变体模型中的白血病负荷(流式细胞术)和生存情况(Kaplan-Meier曲线);(3)临床样本:检测原代细胞的药物反应(CellTiter-Glo法)和PDX模型的疗效。特异性数据:西曲替尼对FLT3-ITD-F691L细胞的IC50为2.1 nM(n=3,P<0.001),吉瑞替尼无抑制;敏感性数据:西曲替尼对FLT3-ITD原代细胞的抑制率为75%-85%(n=3,P<0.01),显著高于其他药物。
核心成果提炼:
1. FLT3-ITD是西曲替尼的有效Biomarker:西曲替尼通过抑制FLT3信号通路发挥抗AML作用,对FLT3-ITD细胞的抑制率显著高于FLT3-WT细胞(P<0.001)。
2. FLT3-ITD-F691L是西曲替尼的特异性Biomarker:西曲替尼可克服该突变介导的吉瑞替尼耐药,其风险比(HR)为0.25(P=0.001),为临床耐药患者提供了新的治疗选择。
3. 细胞因子介导的耐药不影响西曲替尼疗效:西曲替尼通过持续抑制AKT/ERK通路,克服FL/FGF2介导的耐药,其对FL处理后细胞的IC50仅轻度升高(从0.556 nM升至0.8 nM,n=3,P>0.05)。
本研究首次发现FLT3-ITD-F691L可作为西曲替尼的响应Biomarker,为临床筛选西曲替尼的适用患者提供了直接依据,同时证实西曲替尼是一种可克服吉瑞替尼耐药的新型FLT3抑制剂,具有重要的临床转化价值。