Background
The human leukocyte antigen (HLA) genes, exhibiting significant genetic diversity, are associated with susceptibility to various clinical diseases and diverse in drug responses. High costs of HLA sequencing and the population-specific architecture of this genetic region necessitate the establishment of a population-specific HLA imputation reference panel. Moreover, there is a lack of understanding about the genetic and phenotypic landscape of HLA variations within the Taiwanese population.
Conclusions
We successfully delivered the HLA imputation for 59,448 Taiwanese subjects and characterized the genetic and phenotypic landscapes of the HLA variations. In addition, we quantified the estimated prevalence of the ADR-related HLA alleles in the Taiwanese population. The developed HLA imputation reference panel could be used for estimation of population HLA allele frequencies, which can facilitate further studies in the role of HLA variants in a wider range of phenotypes in the population.
Methods
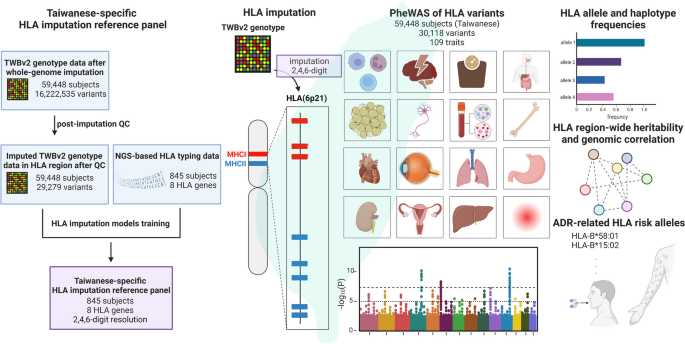
We created models for a Taiwanese-specific HLA imputation reference panel. These models were trained with the array genotype data and HLA sequencing data from 845 Taiwanese subjects. HLA imputation was applied for 59,448 Taiwanese subjects to characterize the HLA allele and haplotype frequencies. Additionally, a phenome-wide association study (PheWAS) was conducted to identify the phenotypes associated with HLA variations. The association of the biallelic HLA variants with the binary and quantitative traits were evaluated with additive logistic and linear regression models, respectively. Furthermore, an omnibus test with likelihood-ratio test was applied for each HLA amino acid position in the multiallelic HLA amino acid polymorphisms to compare the difference between a fitted model and a null model following a χ2 distribution of n-1 degree of freedom at a position with n residues. Finally, we estimated the prevalence of adverse drug reactions (ADR)-related HLA alleles in the Taiwanese population.
Results
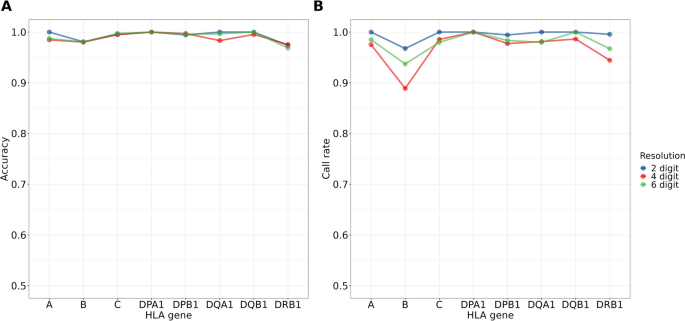
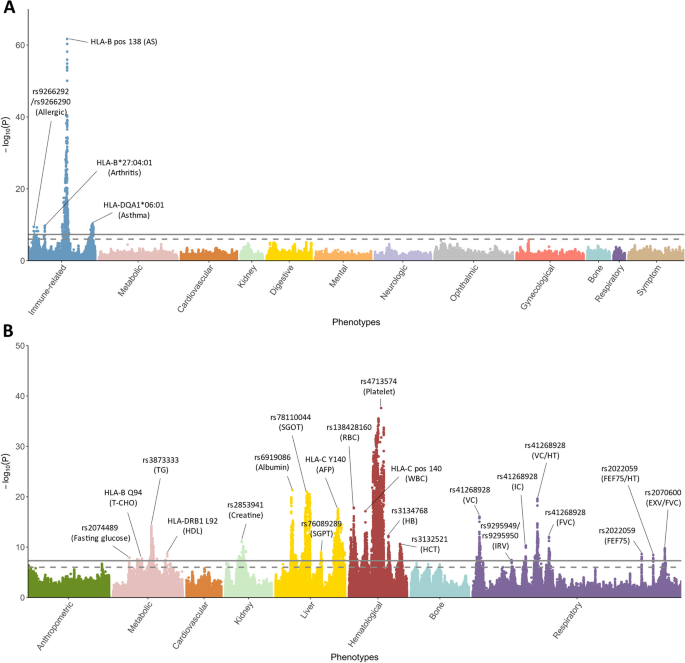
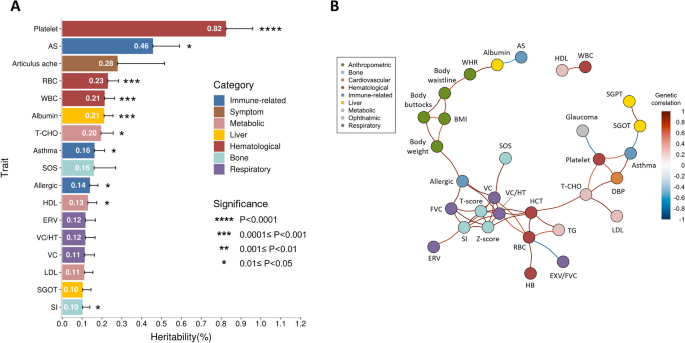
In this study, the reference panel models displayed remarkable accuracy, with averages of 99.3%, 98.9%, and 99.1% for 2-, 4-, 6-digit alleles of the eight classical HLA genes, respectively. For PheWAS, a total of 18,136 significant associations with HLA variants across 26 phenotypes are identified (p < 5×10-8), highlighting the pleiotropy feature of the HLA region. Among the independent signals, 15 are novel, including the association of HLA-B pos 138 variation with ankylosing spondylitis (AS), and rs9266290 and rs9266292 with allergy. Through an analysis spanning the entire HLA region, we identified clusters of phenotype correlations. Finally, the carriers of pharmacogenomic related HLA alleles, including HLA-C*01:02 (35.86%), HLA-B*58:01 (20.9%), and HLA-B*15:02 (8.38%), were characterized in the Taiwanese general population. Conclusions: We successfully delivered the HLA imputation for 59,448 Taiwanese subjects and characterized the genetic and phenotypic landscapes of the HLA variations. In addition, we quantified the estimated prevalence of the ADR-related HLA alleles in the Taiwanese population. The developed HLA imputation reference panel could be used for estimation of population HLA allele frequencies, which can facilitate further studies in the role of HLA variants in a wider range of phenotypes in the population.