1. 领域背景与文献引入
文献英文标题:Modeling gene expression using chromatin features in various cellular contexts;发表期刊:Genome Biology;影响因子:10.309(2012年);研究领域:表观遗传学与基因表达调控。
表观遗传学领域中,组蛋白修饰作为关键的表观调控标记,其与基因表达的关联研究自20世纪末以来持续推进。早期研究通过候选基因验证发现特定组蛋白修饰与基因表达的关联,但无法反映组蛋白修饰的组合调控模式;2000年后,高通量染色质免疫共沉淀测序(ChIP-seq)技术推动了全基因组组蛋白修饰的系统分析,证实组蛋白修饰组合模式与基因表达的关联,但此类研究多局限于单一细胞系,未涉及不同细胞组分与RNA类型的比较。随着ENCODE项目的启动,人类基因组功能元件数据大量产出,涵盖7种人类细胞系的11种组蛋白修饰、1种组蛋白变体及DNase I超敏感位点的全基因组图谱,同时包含多种技术测量的不同细胞组分RNA表达数据,为突破现有研究局限性提供了契机。本研究旨在利用ENCODE多维度数据,构建定量模型揭示不同细胞环境下染色质特征与基因表达的精准关联,填补领域内多细胞系、多RNA类型、多细胞组分分析的空白,为表观遗传调控基因表达的机制提供新视角。
2. 文献综述解析
作者对领域内现有研究的分类维度主要基于研究的系统性与数据覆盖范围,分为早期单一修饰分析、中期系统组蛋白修饰分析、近期定量模型构建三个阶段,重点指出各阶段研究的局限性与未解决问题。
早期研究多聚焦于单个组蛋白修饰的功能解析,通过候选基因验证发现特定组蛋白修饰与基因表达的关联,此类研究的优势是明确了单个标记的调控作用,但无法反映组蛋白修饰的组合调控模式。中期研究借助ChIP-seq技术实现了全基因组组蛋白修饰的系统分析,例如Wang等在人CD4+ T细胞中解析了39种组蛋白修饰的组合模式,证实组蛋白乙酰化与基因表达正相关,此类研究的技术优势是实现了高通量、全基因组的分析,但局限性在于仅针对单一细胞系,未涉及不同细胞系、RNA类型及细胞组分的比较。近期研究开始构建定量模型预测基因表达,如Karlić等利用线性回归模型实现了组蛋白修饰对基因表达的预测,Cheng等将线虫数据的模型应用于人类细胞系,此类模型提升了关联分析的定量精度,但仍缺乏多细胞系、多RNA类型的验证,且未区分基因表达的“开关”状态与表达水平的调控机制。本研究的创新价值在于,首次利用ENCODE项目的多维度数据,构建“分类-回归”两步模型,同时分析不同基因表达测量技术、细胞系、染色质特征类别、CpG启动子类型、RNA类型及细胞组分对预测性能的影响,不仅证实了组蛋白修饰与基因表达的关联在多细胞系中保守,还揭示了转录起始与延伸对应不同染色质特征的预测特异性,以及不同RNA类型与细胞组分的调控差异,弥补了现有研究的局限性。
3. 研究思路总结与详细解析
本研究的整体目标是揭示染色质特征在多种细胞环境下对基因表达的定量调控规律,核心科学问题包括染色质特征如何在不同细胞系中保守预测基因表达、不同基因表达测量技术与细胞组分是否影响预测性能、不同功能类别的染色质特征对转录起始与延伸的预测差异,技术路线遵循“数据获取-预处理-模型构建-多维度验证-结论”的闭环逻辑,通过两步模型(随机森林分类+回归)实现基因表达状态与水平的精准预测。
3.1 数据集获取与预处理
实验目的是获取标准化、可比较的染色质特征与基因表达数据集,为后续模型构建奠定基础。方法细节上,研究人员从ENCODE项目获取7种人类细胞系(K562、GM12878、H1-hESC等)的11种组蛋白修饰、1种组蛋白变体(H2A.Z)及DNase I超敏感位点的全基因组ChIP-seq/DNase-seq数据,同时获取帽分析基因表达(CAGE)、RNA配对末端标签测序(RNA-PET)、RNA测序(RNA-seq)三种技术测量的全细胞、核、胞质组分的PolyA+与PolyA- RNA表达数据;对生物学重复样本取平均值合并,选择每个基因中表达水平最高的转录本作为代表性转录本以避免偏差;对长度超过4100 bp的转录本进行延伸与分箱,通过相关性分析选择与基因表达关联最强的分箱(bestbin),并优化伪计数以解决log2(0)的问题。结果解读显示,bestbin的选择有效捕获了不同染色质特征的信号峰值,例如H3K4me3的bestbin集中在转录起始位点(TSS)附近,H3K36me3的bestbin集中在基因体3"端,为后续模型提供了精准的染色质特征输入。产品关联:文献未提及具体实验产品,领域常规使用ChIP-seq抗体、高通量测序平台等。
3.2 两步预测模型构建
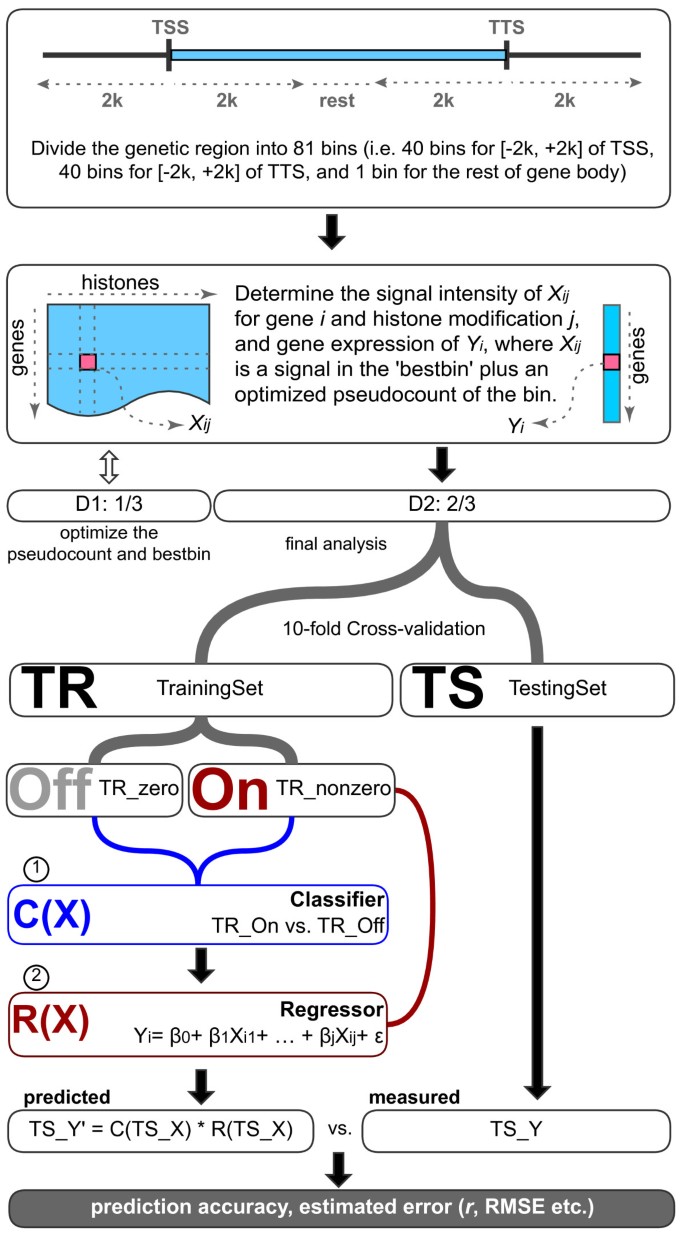
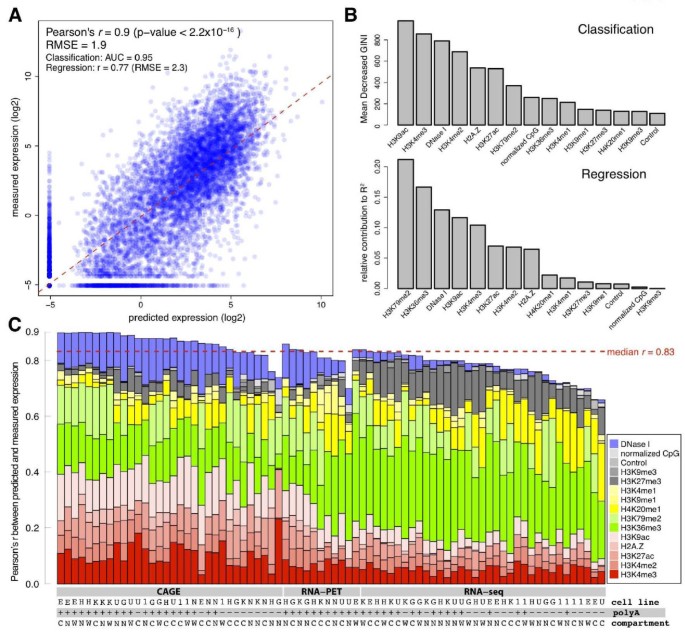
实验目的是构建能够同时区分基因表达状态与预测表达水平的定量模型,解决直接回归分析中大量零表达值的偏差问题。方法细节上,研究人员采用两步模型:第一步利用随机森林分类器,基于染色质特征将基因分为“表达(on)”与“不表达(off)”两类;第二步对“表达”的基因,分别采用线性回归、多元自适应回归样条(MARS)、随机森林回归三种方法预测表达水平;将分类模型的输出(1表示表达,0表示不表达)与回归模型的输出相乘,得到最终的预测表达值;通过十折交叉验证评估模型性能,其中分类模型的性能用受试者工作特征曲线下面积(AUC)评估,回归模型的性能用皮尔逊相关系数(PCC)与均方根误差(RMSE)评估。结果解读显示,两步模型的性能显著优于单一回归模型,例如K562细胞系中CAGE测量的PolyA+胞质RNA数据,两步模型的PCC r=0.9(P<2.2×10^-16),分类模型的AUC=0.95,回归模型的PCC r=0.77、RMSE=2.3;特征重要性分析显示,H3K9乙酰化(H3K9ac)、H3K4me3与DNase I超敏感位点是区分表达状态的关键特征,H3K79二甲基化(H3K79me2)、H3K36me3与DNase I超敏感位点是预测表达水平的关键特征。产品关联:文献未提及具体实验产品,领域常规使用R语言的randomForest、earth等工具包进行模型构建。
3.3 不同基因表达测量技术的比较分析
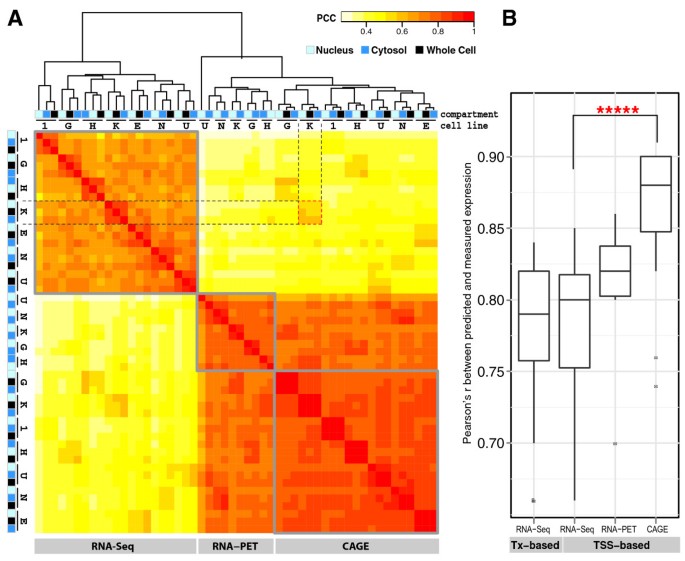
实验目的是比较三种基因表达测量技术(CAGE、RNA-PET、RNA-seq)的可预测性差异,揭示技术特性对染色质特征预测性能的影响。方法细节上,研究人员将两步模型分别应用于三种技术测量的所有数据集,比较各技术的预测PCC分布,并通过配对Wilcoxon检验评估差异的统计学显著性。结果解读显示,三种技术的预测性能均较高(中位数r范围为0.79-0.88),其中CAGE测量的表达可预测性最高,RNA-seq最低(P=3×10^-5);进一步分析显示,基于转录起始位点(TSS)的RNA-seq与基于转录本(Tx)的RNA-seq预测性能相当,排除了多转录本对预测性能的影响。产品关联:文献未提及具体实验产品,领域常规使用CAGE文库构建试剂盒、RNA-seq文库构建试剂盒等。
3.4 不同细胞系的模型适用性分析
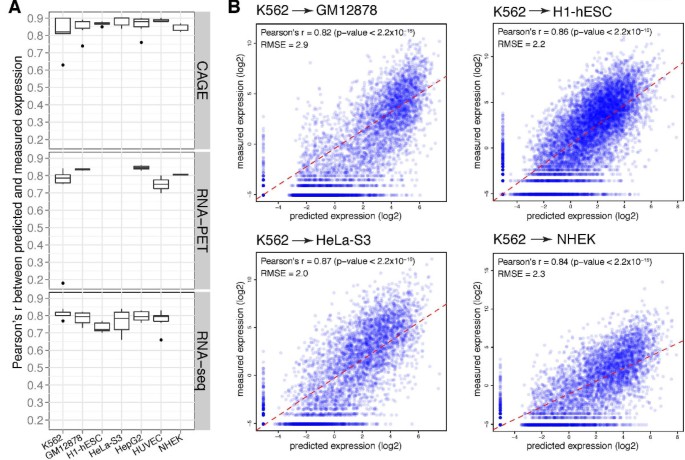
实验目的是验证模型在不同细胞系中的通用性,揭示染色质特征与基因表达的关联是否具有细胞系特异性。方法细节上,研究人员将模型应用于7种人类细胞系的数据集,比较各细胞系的预测性能,并将K562细胞系的模型应用于其他细胞系,评估跨细胞系预测的性能。结果解读显示,模型在所有细胞系中均表现良好(平均r=0.8),其中H1-hESC细胞系的RNA-seq预测性能显著低于其他细胞系(P=0.02-0.07),推测可能是由于干细胞中存在大量转录暂停基因,CAGE可捕获转录起始但RNA-seq无法有效捕获延伸过程;跨细胞系预测的性能较高,例如K562模型预测GM12878的r=0.82,表明染色质特征与基因表达的关联具有保守性。产品关联:文献未提及具体实验产品,领域常规使用细胞培养试剂、流式细胞仪等进行细胞系培养与鉴定。
3.5 不同染色质特征类别的预测贡献分析
实验目的是分析不同功能类别的染色质特征对基因表达的预测贡献,揭示转录起始与延伸过程的染色质调控差异。方法细节上,研究人员将染色质特征分为四类:启动子标记(H3K4me2、H3K4me3、H2A.Z、H3K9ac、H3K27ac)、结构标记(H3K36me3、H3K79me2)、抑制标记(H3K27me3、H3K9me3)、远端/其他标记(H3K4me1、H4K20me1、H3K9me1),分别评估各类别及组合的预测性能。结果解读显示,对于TSS-based的表达数据(CAGE、RNA-PET),启动子标记的预测性最高;对于Tx-based的表达数据(RNA-seq),结构标记的预测性最高,证实启动子标记主要调控转录起始,结构标记主要调控转录延伸;DNase I超敏感位点的预测性低于启动子标记,表明开放染色质是转录的基础,但组蛋白修饰在精细调控表达水平中发挥更重要的作用。产品关联:文献未提及具体实验产品,领域常规使用ChIP-seq特异性抗体检测组蛋白修饰。
3.6 不同CpG启动子基因的预测差异分析
实验目的是比较高CpG启动子(HCP)与低CpG启动子(LCP)基因的可预测性差异,揭示启动子类型对染色质调控的影响。方法细节上,研究人员根据启动子区域的标准化CpG含量将基因分为HCP(CpG>0.4)与LCP(CpG≤0.4)两组,分别应用模型并比较预测性能,同时分析两组基因的特征重要性差异。结果解读显示,HCP基因的可预测性显著高于LCP基因(中位数r=0.8 vs 0.66,P=2.19×10^-14);特征重要性分析显示,HCP基因中结构标记对RNA-seq的预测性最高,LCP基因中启动子标记对CAGE/RNA-PET的预测性更高,推测LCP基因的转录起始与延伸调控可能存在解偶联。产品关联:文献未提及具体实验产品,领域常规使用亚硫酸氢盐测序技术分析CpG甲基化水平。
3.7 不同RNA类型与细胞组分的预测差异分析
实验目的是分析不同RNA类型(PolyA+ vs PolyA-)与细胞组分(胞质、核、全细胞)的可预测性差异,揭示RNA加工与转运过程的调控机制。方法细节上,研究人员比较PolyA+与PolyA- RNA的预测性能,以及不同细胞组分中RNA的预测性能,通过配对Wilcoxon检验评估差异的统计学显著性。结果解读显示,PolyA+ RNA的可预测性显著高于PolyA- RNA(P<2.2×10^-16),表明PolyA- RNA的调控机制可能与PolyA+ RNA不同;对于RNA-seq数据,PolyA+胞质RNA的预测性显著高于核RNA(P=0.01),而PolyA-核RNA的预测性显著高于胞质RNA(P=0.03),推测这与RNA的稳定性与转运过程相关,PolyA+胞质RNA与PolyA-核RNA是各类型中的主要群体,其丰度主要由转录调控决定,而次要群体的丰度可能受降解过程影响,无法通过染色质特征有效预测。产品关联:文献未提及具体实验产品,领域常规使用PolyA富集试剂盒、细胞组分分离试剂盒等进行RNA的分离与富集。
4. Biomarker研究及发现成果解析
本研究中涉及的Biomarker为具有基因表达预测能力的染色质特征,按功能可分为转录起始预测Biomarker、转录延伸预测Biomarker、启动子类型相关Biomarker三类,其筛选与验证基于ENCODE多维度数据与两步模型的系统分析。
Biomarker定位方面,转录起始预测Biomarker包括H3K4me3、H3K9ac、H2A.Z等启动子标记,筛选逻辑是通过特征重要性分析发现其对TSS-based表达数据(CAGE、RNA-PET)的预测性最高,验证逻辑是在7种细胞系中均表现出高预测性能;转录延伸预测Biomarker包括H3K36me3、H3K79me2等结构标记,筛选逻辑是其对Tx-based表达数据(RNA-seq)的预测性最高,验证逻辑是在多细胞系中保守;启动子类型相关Biomarker为启动子区域的CpG含量,筛选逻辑是HCP基因的可预测性显著高于LCP基因,验证逻辑是在所有实验数据中均存在此差异。研究过程详述显示,这些Biomarker的来源为7种人类细胞系的全基因组染色质特征数据,验证方法为两步模型的十折交叉验证,其中转录起始Biomarker的预测AUC可达0.95,转录延伸Biomarker的预测PCC r可达0.77,CpG启动子类型的预测差异P=2.19×10^-14。核心成果提炼显示,转录起始Biomarker可精准预测基因的表达状态(开关),转录延伸Biomarker可精准预测基因的表达水平,CpG启动子类型可预测基因表达的可调控性;本研究首次揭示了不同染色质特征在转录起始与延伸过程中的特异性调控作用,以及不同RNA类型与细胞组分的调控差异,为表观遗传调控的精准解析提供了新的Biomarker体系,相关统计学结果均具有显著的统计学意义(P<0.05)。