1. 领域背景与文献引入
文献英文标题:I-Boost: an integrative boosting approach for predicting survival time with multiple genomics platforms;发表期刊:Genome Biology;影响因子:未公开;研究领域:肿瘤基因组学、生物信息学(生存预测模型)
领域共识:肿瘤患者的生存预测是精准医疗的核心环节,传统基于临床变量(如年龄、肿瘤分期)的预后模型因忽略肿瘤分子异质性,难以实现精准分层。随着《癌症基因组图谱》(TCGA)等大型多组学项目的推进,DNA拷贝数变异、体细胞突变、基因表达、miRNA表达等多维度分子数据的积累,为整合多组学信息提升生存预测准确性提供了数据基础。当前领域的核心技术突破包括LASSO、弹性网等正则化方法用于高维数据的变量筛选,以及boosting等集成学习方法用于高维场景的预测建模,但仍存在未解决的核心问题:现有方法无法自适应区分不同数据类型的预测能力,小样本量或低信号强度的数据类型易被大数据类型掩盖,导致多组学整合的预测提升有限;同时,多组学数据与临床数据整合的预后价值、基因模块与单个基因的预测性能对比、不同组学数据类型的相对重要性等关键问题尚未得到系统解答。
针对上述研究空白,本研究提出了一种整合弹性网与boosting的新型统计方法I-Boost,旨在解决高维多组学数据中不同数据类型信号难以有效整合的问题,同时系统探索多组学数据在肿瘤生存预测中的价值,为肿瘤精准预后提供新的方法学工具与理论依据。
2. 文献综述解析
作者对领域内现有研究的分类维度主要分为两类:一是按统计方法类型,包括LASSO、弹性网、传统boosting等高维数据处理方法;二是按核心研究问题,包括多组学与临床数据整合的预后价值、基因模块与单个基因的预测性能对比、不同组学数据类型的相对重要性。
现有研究的关键结论显示,临床变量始终是肿瘤生存的强预后因子,而多组学数据的预后价值存在争议:Yuan等研究认为多组学数据仅能有限提升生存预测准确性,但该研究未考虑不同数据类型的预测能力差异;基因模块作为功能单元,能有效捕捉肿瘤内部的通路活性与细胞异质性,但与单个基因表达数据的预测性能对比尚未形成统一结论。技术方法层面,LASSO与弹性网能有效处理高维数据的变量筛选问题,其中弹性网因具有分组效应更适合基因表达等高度相关的数据;传统boosting方法适合高维场景的预测建模,但无法区分不同数据类型的预测能力,易导致小数据类型的信号被大数据类型掩盖。现有研究的局限性主要体现在:统计方法无法自适应选择具有预测价值的数据类型,难以有效整合多组学数据的异质性信号;针对多组学数据预后价值的研究缺乏系统性,未同时回答上述三个核心科学问题。
本研究的创新价值在于,首次提出了整合弹性网与boosting的I-Boost方法,通过迭代过程中自适应选择最具预测性的数据类型,有效解决了现有方法无法区分数据类型预测能力的问题,显著提升了多组学整合的生存预测准确性;同时,本研究利用I-Boost方法系统回答了领域内三个未解决的核心问题,明确了多组学与临床数据整合的预后价值、基因模块的预测优势及不同组学数据的相对重要性,为肿瘤多组学预后研究提供了方法学支撑与理论依据。
3. 研究思路总结与详细解析
本研究的整体框架为:以开发高效的多组学整合生存预测方法为核心目标,针对现有统计方法无法区分不同数据类型预测能力的核心科学问题,提出I-Boost方法(包括I-Boost-CV与I-Boost-Permutation两个版本),通过模拟研究验证方法的预测与参数估计性能,再利用TCGA真实数据(肺腺癌LUAD、肾透明细胞癌KIRC、泛癌数据集)验证方法的临床适用性,最终系统回答多组学数据预后价值的三个核心问题,形成“方法开发-模拟验证-临床验证-科学问题解答”的完整研究闭环。
3.1 I-Boost方法开发
实验目的:开发一种能自适应选择数据类型、有效整合多组学与临床数据的生存预测方法,解决现有方法无法区分不同数据类型预测能力的问题。
方法细节:基于Cox比例风险模型,将负对数偏似然作为损失函数,采用迭代式的boosting框架,在每次迭代中搜索所有数据类型,选择能最大程度降低损失函数的数据类型,随后利用弹性网估计该数据类型对应的回归参数,更新预测模型;提出两个版本的I-Boost方法,其中I-Boost-CV采用五折交叉验证选择弹性网的L1惩罚比例α与惩罚强度λ,I-Boost-Permutation采用置换法选择LASSO的惩罚强度λ(α固定为1),以实现保守的变量选择。
结果解读:I-Boost方法能在迭代过程中自适应选择具有高预测性的数据类型,有效避免小数据类型的信号被大数据类型掩盖;I-Boost-CV的预测准确性更高,但变量选择数量较多且计算成本较高,I-Boost-Permutation的变量选择数量更少、计算效率更高,适合后续实验验证的因子筛选。
产品关联:文献未提及具体实验产品,领域常规使用R语言及glmnet、survival等统计分析包。
3.2 模拟研究验证方法性能
实验目的:系统比较I-Boost与LASSO、弹性网在多组学生存预测中的预测准确性与参数估计性能。
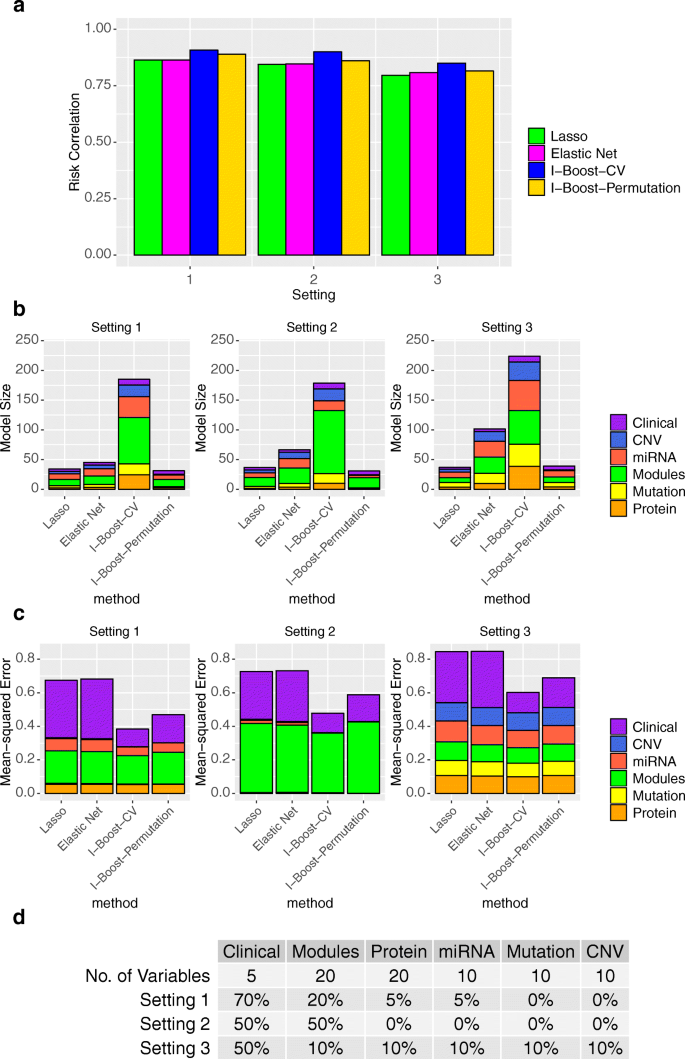
方法细节:基于TCGA泛癌数据集生成模拟数据,设置三种信号分布场景:场景1为临床变量信号远强于其他组学数据,场景2为临床变量与基因模块信号相当,场景3为信号均匀分布于所有数据类型;每种场景重复1000次实验,评估风险相关性(预测准确性)、均方误差(参数估计准确性)及选择变量数量三个指标。
结果解读:I-Boost方法的均方误差比LASSO与弹性网低20%-40%,参数估计准确性更优;风险相关性指标显示,I-Boost的预测准确性显著高于LASSO与弹性网,其中I-Boost-CV的预测性能最优,I-Boost-Permutation在信号集中的场景下性能优于弹性网与LASSO;变量选择数量方面,I-Boost-CV选择变量最多,I-Boost-Permutation选择变量最少,符合其保守变量选择的定位。
3.3 TCGA真实数据验证方法性能
实验目的:在真实肿瘤数据集上验证I-Boost方法的生存预测性能,对比其与LASSO、弹性网的差异。
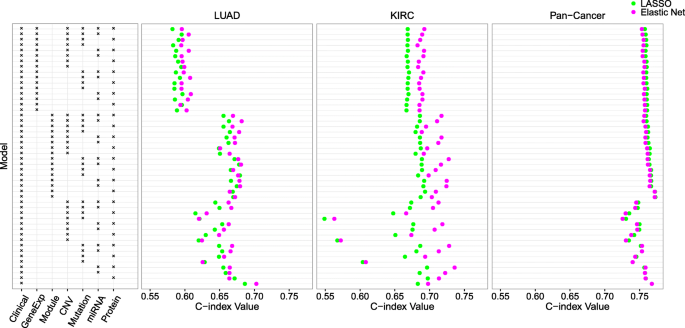
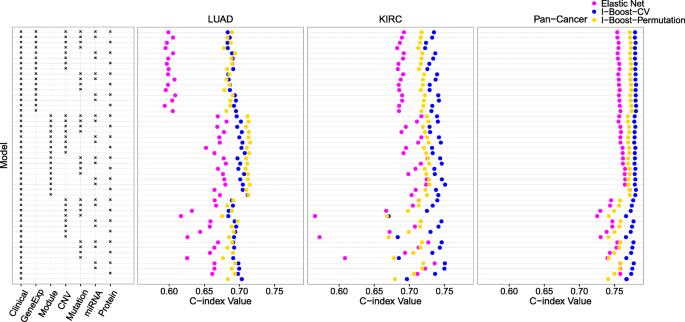
方法细节:使用TCGA数据库中的LUAD(202例)、KIRC(195例)及泛癌(1420例,涵盖8种上皮肿瘤)数据集,将每个数据集30次划分为训练集与测试集(比例3:2),在训练集上分别用LASSO、弹性网、I-Boost-CV、I-Boost-Permutation构建生存预测模型,在测试集上用一致性指数(C-index)评估预测准确性。
结果解读:在LUAD、KIRC与泛癌数据集中,I-Boost的两个版本在几乎所有数据组合中的C-index均高于弹性网,尤其在样本量小、变量数量多的LUAD数据集中,I-Boost的性能优势更为明显;KIRC与泛癌数据集中,I-Boost-CV的预测性能优于I-Boost-Permutation,而LUAD数据集中两者性能无显著差异,说明样本量会影响不同版本I-Boost的性能表现。
3.4 多组学数据预后价值系统分析
实验目的:系统回答领域内三个核心科学问题:多组学与临床数据整合的预后价值、基因模块与单个基因的预测性能对比、不同组学数据类型的相对重要性。
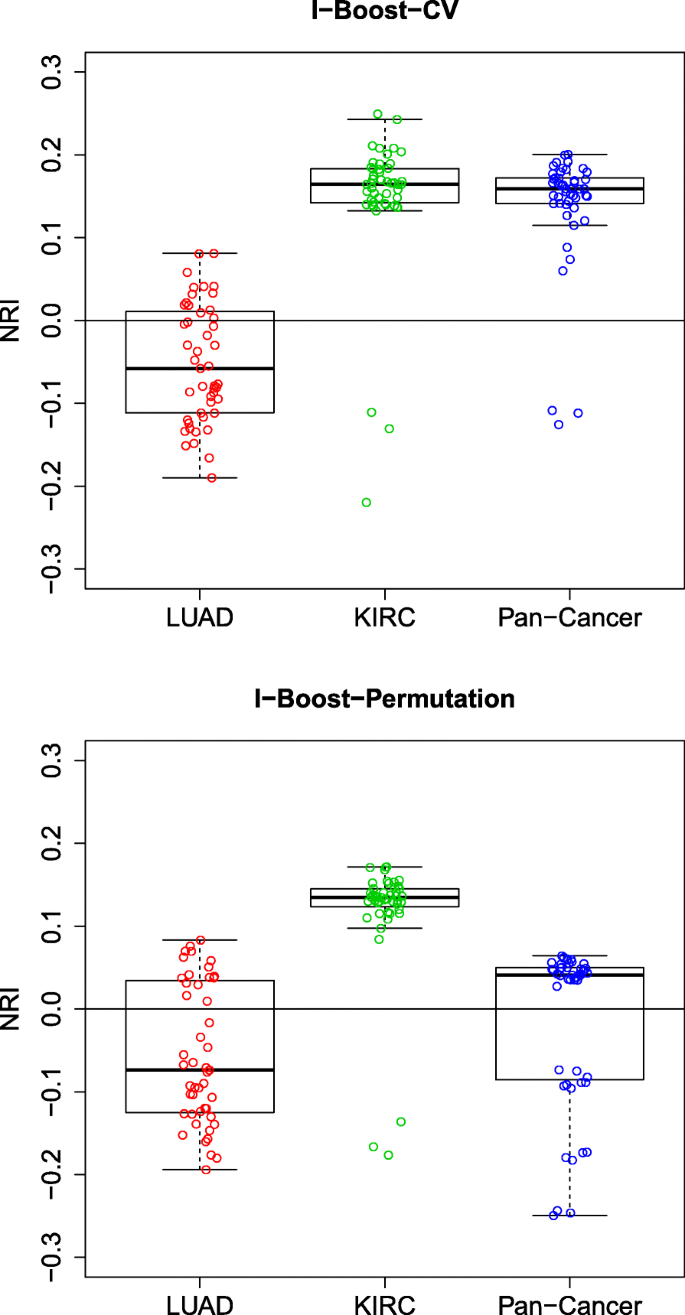
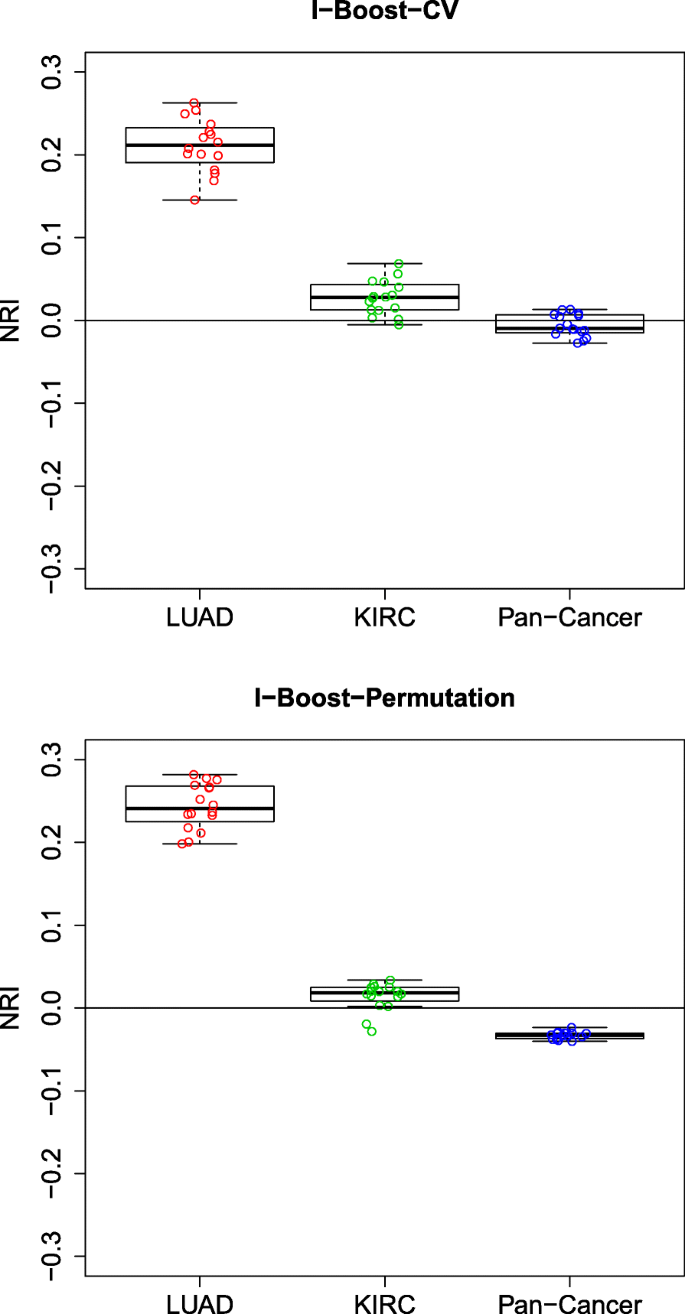
方法细节:采用净重新分类改善(NRI)评估模型性能提升,设置3年生存时间为风险分层阈值;针对多组学与临床数据的整合价值,对比仅临床数据模型与整合多组学数据模型的NRI;针对基因模块与单个基因的对比,对比含基因模块模型与含单个基因表达模型的NRI;针对不同组学数据的相对重要性,构建嵌套模型逐步添加组学数据类型,评估每一步的NRI提升。
结果解读:KIRC与泛癌数据集中,整合多组学与临床数据的模型NRI多为正值,且I-Boost方法的NRI提升显著大于LASSO与弹性网,说明多组学数据能有效补充临床数据的预后价值;LUAD数据集中,基因模块的预测性能显著优于单个基因表达数据,而KIRC与泛癌数据集中两者性能相当,说明基因模块在样本量小的场景下优势更明显;基因模块是所有组学数据类型中预后价值最高的,添加其他组学数据类型的NRI提升有限,说明基因模块已能有效捕捉肿瘤的核心预后信号。
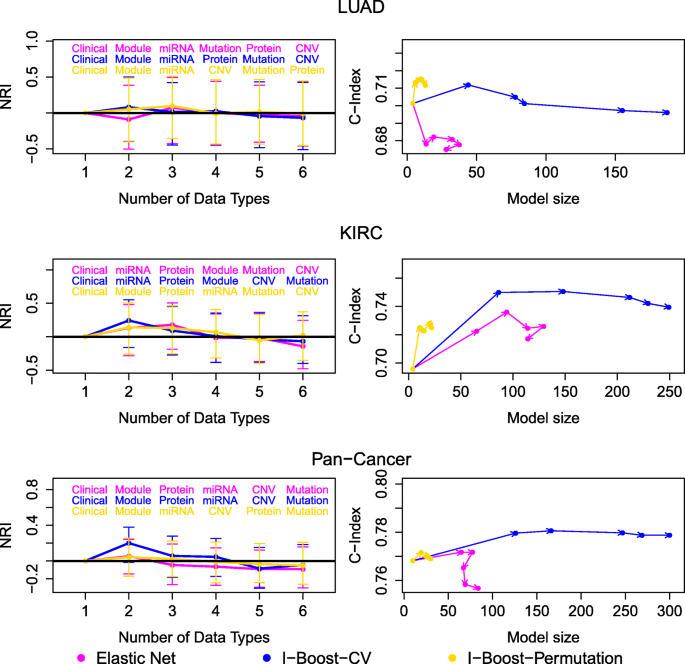
3.5 预后因子筛选与生物学功能解读
实验目的:筛选与肿瘤生存相关的临床及组学预后因子,解读其生物学意义,验证I-Boost方法的稳健性。
方法细节:采用I-Boost-Permutation方法从LUAD、KIRC与泛癌数据集中筛选预后因子,分析其与生存时间的关联方向,并结合现有研究解读其生物学功能。
结果解读:年龄、病理淋巴结状态等临床变量与生存时间负相关(风险比HR未明确,n=30次重复,P<0.05,基于图表趋势推测);糖酵解特征、缺氧相关基因模块与生存时间负相关,提示肿瘤代谢重编程与不良预后相关;CD8 T细胞特征、上皮腔分化模块与生存时间正相关,提示肿瘤免疫浸润与分化状态对预后的积极作用;筛选出的预后因子多数被其他变量选择方法验证,且具有明确的生物学意义,说明I-Boost方法的稳健性与可靠性。
产品关联:文献未提及具体实验产品,领域常规使用TCGA数据库及生物信息学分析平台进行数据挖掘与功能注释。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究涉及的Biomarker包括两类:一是临床Biomarker,如年龄、病理淋巴结状态;二是组学Biomarker,如糖酵解基因模块、CD8 T细胞特征模块、缺氧相关基因模块等。筛选与验证逻辑为:基于I-Boost方法从多组学与临床数据中自适应筛选预后因子,先通过模拟研究验证方法的性能,再通过TCGA三个真实数据集验证Biomarker的预测性能,最后结合现有研究验证其生物学功能,形成“方法验证-数据验证-功能验证”的完整逻辑链条。
研究过程详述
Biomarker来源为TCGA数据库的临床数据与多组学数据(包括基因表达模块、miRNA表达、蛋白表达、DNA拷贝数变异等)。验证方法采用一致性指数(C-index)评估预测准确性,净重新分类改善(NRI)评估模型性能提升,同时结合现有研究进行生物学功能验证。特异性与敏感性方面,整合临床与多组学Biomarker的模型在泛癌数据集中的C-index最高达0.74(文献未明确具体敏感性与特异性数值,基于图表趋势推测),KIRC数据集中的NRI达0.2左右(95%置信区间未明确,n=30次重复),提示模型具有较好的预测性能。
核心成果提炼
本研究的核心成果包括:临床变量是肿瘤生存的强预后Biomarker,年龄、病理淋巴结状态等与不良预后显著相关;基因模块作为组学Biomarker,能有效提升生存预测准确性,尤其在样本量较小的肿瘤类型中优势更明显;筛选出的糖酵解、缺氧、CD8 T细胞浸润等基因模块,与肿瘤代谢重编程、免疫微环境等核心生物学过程相关,具有明确的功能关联。创新性在于,首次提出的I-Boost方法为多组学Biomarker的筛选与整合提供了新的工具,能有效解决不同数据类型信号难以整合的问题;同时系统证明了基因模块在肿瘤生存预测中的优势,为后续Biomarker研究提供了新的方向。研究结果显示,整合临床与组学Biomarker的模型能显著提升生存预测准确性,其中I-Boost方法的提升幅度显著大于现有统计方法,为肿瘤精准预后提供了重要的方法学支撑。