1. 领域背景与文献引入
文献英文标题:Benchmarking deconvolution methods using real bulk RNA-seq data identifies matrix cancer-associated fibroblasts as a pan-cancer prognostic marker;发表期刊:Genome Biology;影响因子:未公开;研究领域:肿瘤学、计算生物学(肿瘤微环境细胞解构)
肿瘤微环境的细胞异质性是影响肿瘤进展、治疗响应及患者预后的核心因素,解析细胞亚型组成及功能是肿瘤精准医学的关键方向。领域共识:单细胞RNA测序(scRNA-seq)技术实现了单细胞分辨率的基因表达分析,能精准解析肿瘤微环境中的细胞亚型,但因测序成本高昂,难以应用于大规模临床队列研究;而bulk RNA-seq及芯片数据(如TCGA、GEO数据库)包含丰富的临床信息,样本量庞大,是开展肿瘤预后及机制研究的重要资源,但无法直接解析细胞组成。反卷积方法作为连接两者的桥梁,能从bulk数据中估算细胞类型比例,成为肿瘤微环境研究的核心工具。然而,当前领域存在未解决的核心问题:传统反卷积方法的基准测试依赖伪bulk数据,而伪bulk数据无法模拟真实bulk样本的生物异质性、批次效应及临床样本复杂性,导致方法性能在真实临床场景中不可靠,且现有基准测试缺乏与临床预后相关的评估维度,限制了反卷积方法的临床转化应用。针对这一问题,本研究建立了基于真实bulk RNA-seq数据的反卷积方法基准框架,通过三个临床相关的评估场景筛选可靠方法,并挖掘具有泛癌预后价值的细胞亚型,为肿瘤精准医学提供了可操作的工具和新的预后标志物。
2. 文献综述解析
作者对领域内现有研究的分类维度主要为反卷积方法的基准测试数据类型(伪bulk数据vs真实bulk数据),以及方法性能的评估维度(细胞比例估计准确性vs临床相关指标重现性)。
现有研究的关键结论是反卷积方法能有效从bulk数据中解析肿瘤微环境的细胞组成,为肿瘤异质性研究提供了可行路径;技术方法优势在于伪bulk数据易于构建,可通过混合已知比例的细胞模拟bulk数据,用于初步的方法性能测试;但现有研究存在明显局限性,一是伪bulk数据无法真实反映临床样本的生物异质性和批次效应,导致在伪bulk数据上表现优异的方法在真实bulk数据中性能下降,二是现有基准测试多聚焦于细胞比例的估计准确性,缺乏与临床预后相关的评估维度,无法满足临床转化研究的需求。
本研究的创新价值在于首次建立了基于差异比例(DP)细胞类型和预后相关(PR)细胞类型的真实bulk数据基准框架,从与scRNA-seq的一致性、跨队列重现性、预后相关细胞亚型重现性三个临床相关场景评估反卷积方法,弥补了传统伪bulk基准的局限性;同时,通过可靠的反卷积方法挖掘到基质癌相关成纤维细胞(mCAF)作为泛癌预后标志物,并构建了经典单核细胞+mCAF的联合预后指标,为泛癌预后研究提供了新的生物标志物,其研究结果直接服务于临床转化需求,具有重要的学术价值。
3. 研究思路总结与详细解析
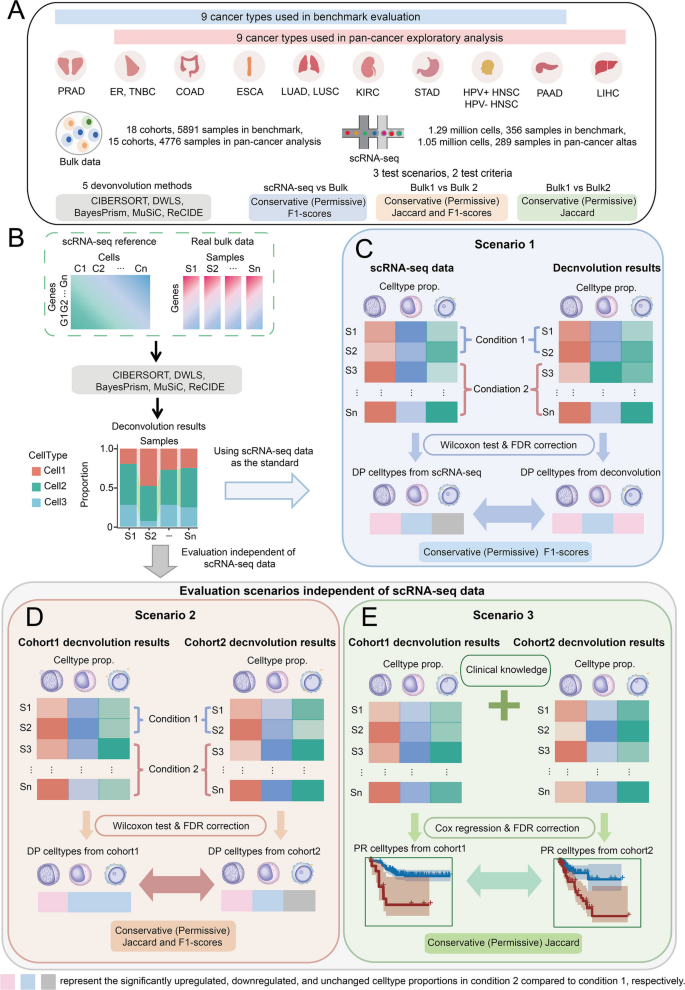
本研究的整体框架为:针对传统反卷积方法基准测试的局限性,提出基于差异比例(DP)和预后相关(PR)细胞类型的真实bulk数据评估思路,构建三个评估场景对五种反卷积方法进行系统评估,筛选出性能最优的ReCIDE和BayesPrism方法;随后基于这两种方法开展泛癌预后分析,识别到mCAF为泛癌预后标志物,并构建联合预后指标进行多队列验证,形成“方法基准测试→可靠方法应用→生物标志物挖掘→临床验证”的完整研究闭环。研究目标是建立真实bulk数据的反卷积方法基准体系,筛选适合临床研究的可靠方法,并挖掘具有泛癌预后价值的细胞亚型;核心科学问题是如何基于真实bulk数据评估反卷积方法的临床可靠性,以及哪些细胞亚型具有跨癌种的预后预测价值。
3.1 基准框架设计与方法筛选
实验目的:解决传统伪bulk基准测试的局限性,建立基于真实bulk数据的反卷积方法评估框架,排除存在偏倚的方法,确定后续评估的方法集合。
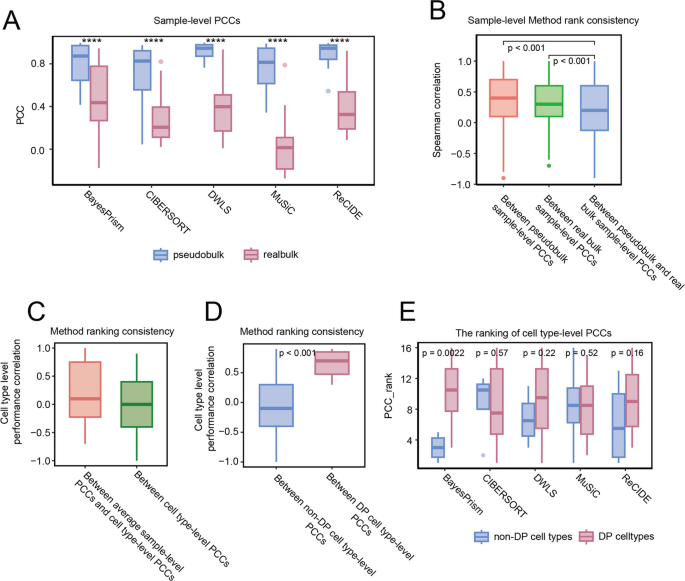
方法细节:首先利用配对的乳腺癌scRNA-seq和bulk RNA-seq数据集(GSE176078),比较伪bulk数据(通过SimBu生成)与真实bulk数据中反卷积方法的性能差异;随后定义差异比例(DP)细胞类型,即通过Wilcoxon检验(FDR<0.2)从scRNA-seq数据中筛选出在疾病条件间比例显著变化的细胞类型;初始选择五种广泛使用的反卷积方法(CIBERSORT、DWLS、MuSiC、Bisque、BayesPrism),并纳入新开发的ReCIDE方法,通过预实验评估方法的参考依赖偏倚,排除存在明显偏倚的Bisque方法。
结果解读:通过GSE176078数据验证发现,伪bulk数据的样本水平Pearson相关系数(PCC)显著高于真实bulk数据,但方法排名在伪bulk与真实bulk数据间的一致性差(p<0.001),说明伪bulk基准无法反映真实场景的方法性能;DP细胞类型在真实bulk数据中的方法排名一致性显著高于非DP细胞类型(p<0.001),且估计准确性更高,证明基于DP细胞类型的评估更具鲁棒性;预实验发现Bisque方法存在参考依赖偏倚,其结果随scRNA-seq参考的细胞比例变化而显著波动,因此被排除,最终确定五种方法(CIBERSORT、DWLS、MuSiC、BayesPrism、ReCIDE)进行后续评估。
产品关联:文献未提及具体实验产品,领域常规使用R语言相关生物信息学包(如Seurat、SimBu、stats等)、测序数据处理软件及高性能计算服务器资源。
3.2 反卷积结果与scRNA-seq的DP细胞类型一致性评估
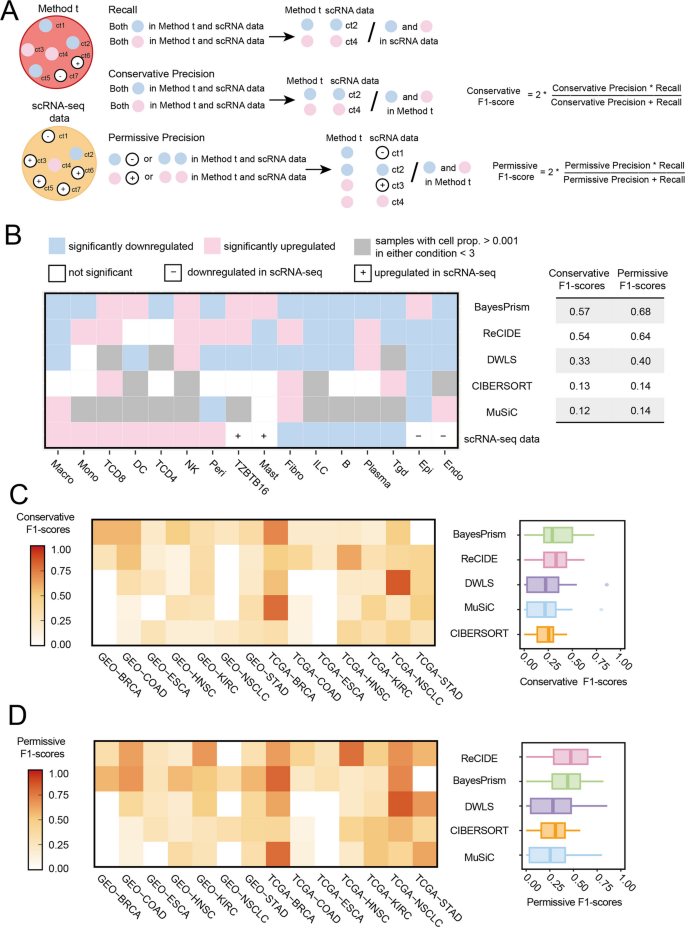
实验目的:评估反卷积方法识别的DP细胞类型与scRNA-seq定义的DP细胞类型的一致性,验证方法的细胞亚型解析准确性。
方法细节:以F1-score为核心评估指标,分为保守和宽松两种标准:保守标准下,精确率为反卷积识别的DP细胞类型中被scRNA-seq验证的比例,召回率为scRNA-seq定义的DP细胞类型中被反卷积识别的比例;宽松标准下,精确率包含趋势一致但未达统计显著的细胞类型。在8种癌症类型中开展分析,排除PRAD(其scRNA-seq数据中无DP细胞类型)。
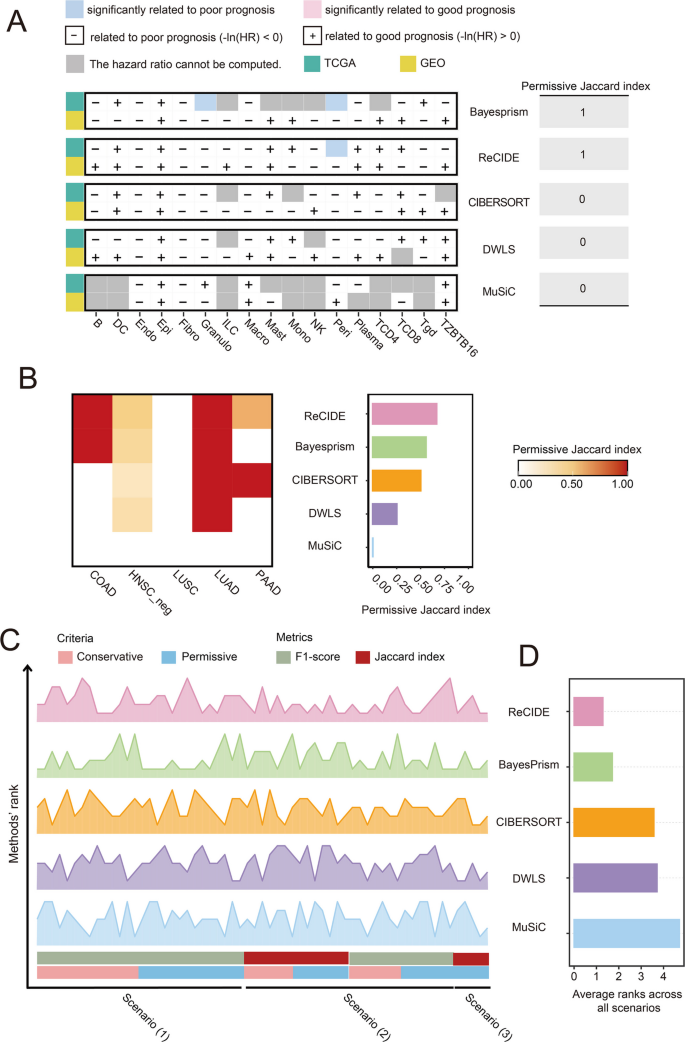
结果解读:在结直肠癌(COAD)队列中,BayesPrism的保守F1-score为0.57(召回率0.67,精确率0.5),ReCIDE为0.54(召回率0.58,精确率0.5),MuSiC仅为0.12,显示出明显的性能差异;跨8种癌症类型的分析显示,BayesPrism和ReCIDE的保守F1-score分别为0.35和0.33,宽松F1-score为0.44和0.46,均显著优于其他方法(DWLS、CIBERSORT、MuSiC);排除丰度<1%的DP细胞类型后,方法排名保持一致,证明评估框架的鲁棒性。
产品关联:文献未提及具体实验产品,领域常规使用R语言的stats、ggpubr包进行统计分析和可视化。
3.3 跨bulk队列的DP细胞类型重现性评估
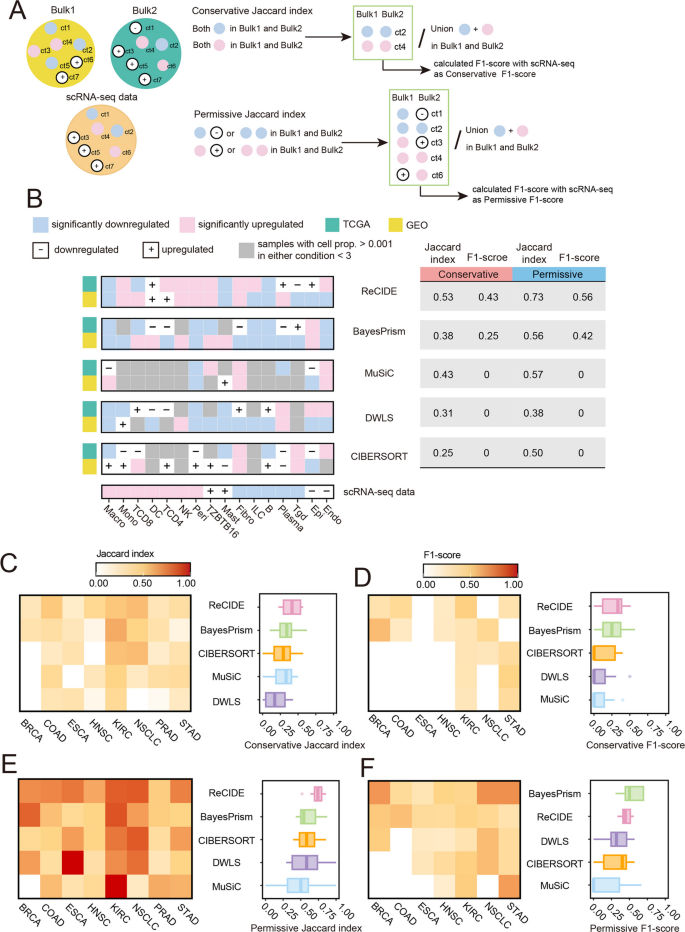
实验目的:评估反卷积方法在独立bulk队列中识别DP细胞类型的重现性,验证方法的稳定性和临床适用性。
方法细节:以Jaccard指数为核心评估指标,分为保守和宽松两种标准:保守标准要求DP细胞类型在两个独立队列(TCGA和GEO)中均达统计显著,宽松标准要求在一个队列中显著且在另一个队列中趋势一致;同时用F1-score评估共享DP细胞类型与scRNA-seq定义的DP细胞类型的一致性。
结果解读:在COAD队列中,ReCIDE的保守Jaccard指数为0.53,BayesPrism为0.38;跨8种癌症类型的分析显示,ReCIDE的保守Jaccard指数为0.41,宽松Jaccard指数为0.73,BayesPrism的保守和宽松Jaccard指数分别为0.34和0.61,均显著优于其他方法;F1-score方面,保守标准下ReCIDE为0.27,BayesPrism为0.26,宽松标准下BayesPrism为0.54,ReCIDE为0.46,显示两种方法各有优势。
产品关联:文献未提及具体实验产品,领域常规使用R语言的stats、pheatmap包进行统计分析和可视化。
3.4 跨队列的预后相关(PR)细胞类型重现性评估
实验目的:评估反卷积方法识别的PR细胞类型在跨队列中的重现性,突出方法的临床相关性。
方法细节:选择5种具有配对生存数据的癌症实体(COAD、HPV-HNSC、LUAD、LUSC、PAAD),通过多变量Cox回归分析识别PR细胞类型;以Jaccard指数评估跨队列的重现性,保守标准要求PR细胞类型在两个队列中均达统计显著,宽松标准要求在一个队列中显著且趋势一致;同时通过调整FDR阈值(0.05→0.10→0.15)验证方法性能的稳定性。
结果解读:保守标准下,无方法能识别到在两个队列中均显著的共享PR细胞类型;宽松标准下,ReCIDE的Jaccard指数为0.67,BayesPrism为0.56,CIBERSORT为0.5,DWLS为0.25,MuSiC未识别到共享PR细胞类型;调整FDR阈值后,ReCIDE和BayesPrism仍保持领先地位,其他方法排名无明显变化,证明两种方法在预后相关细胞亚型识别中的稳定性。
产品关联:文献未提及具体实验产品,领域常规使用R语言的survival、survminer包进行生存分析和可视化。
3.5 泛癌预后分析与联合预后指标验证
实验目的:基于性能最优的反卷积方法,挖掘具有泛癌预后价值的细胞亚型,并构建联合预后指标进行多队列验证,验证其临床应用价值。
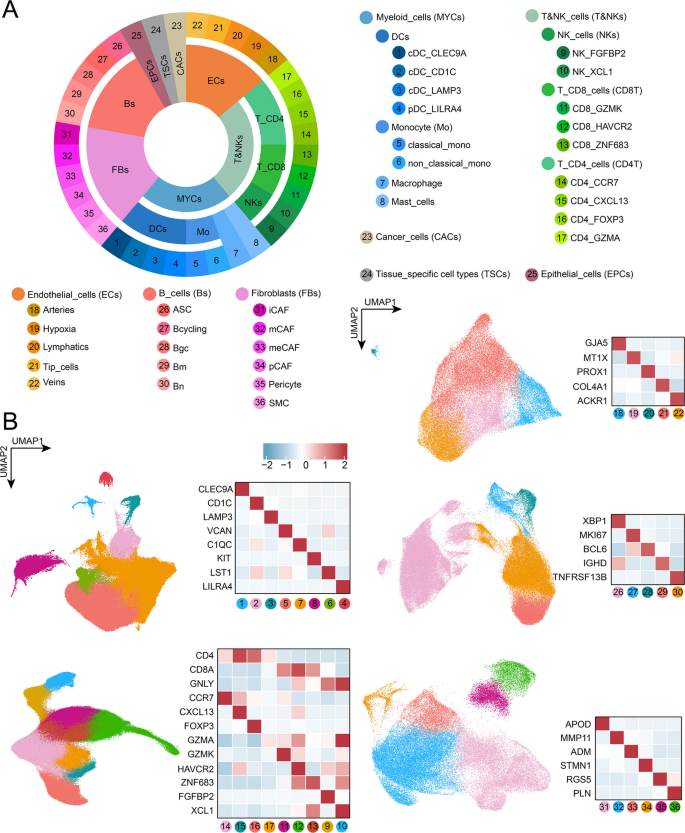
方法细节:首先整合10种癌症类型的scRNA-seq数据,构建包含1.05M细胞的泛癌细胞图谱,通过Seurat包进行细胞聚类和亚型注释,得到33种细胞亚型;随后利用ReCIDE和BayesPrism方法对11种具有生存数据的癌症实体的TCGA队列进行反卷积分析,通过多变量Cox回归分析细胞亚型与患者生存的关联,利用单样本Wilcoxon符号秩检验(FDR校正)筛选具有泛癌预后关联的细胞亚型;基于筛选到的mCAF,构建经典单核细胞比例+mCAF比例的联合预后指标,在5个TCGA队列、5个GEO基准队列及2个额外GEO队列(GSE17536、GSE30219)中验证其预后价值;同时对mCAF的标记基因进行GO富集分析,探索其功能机制。
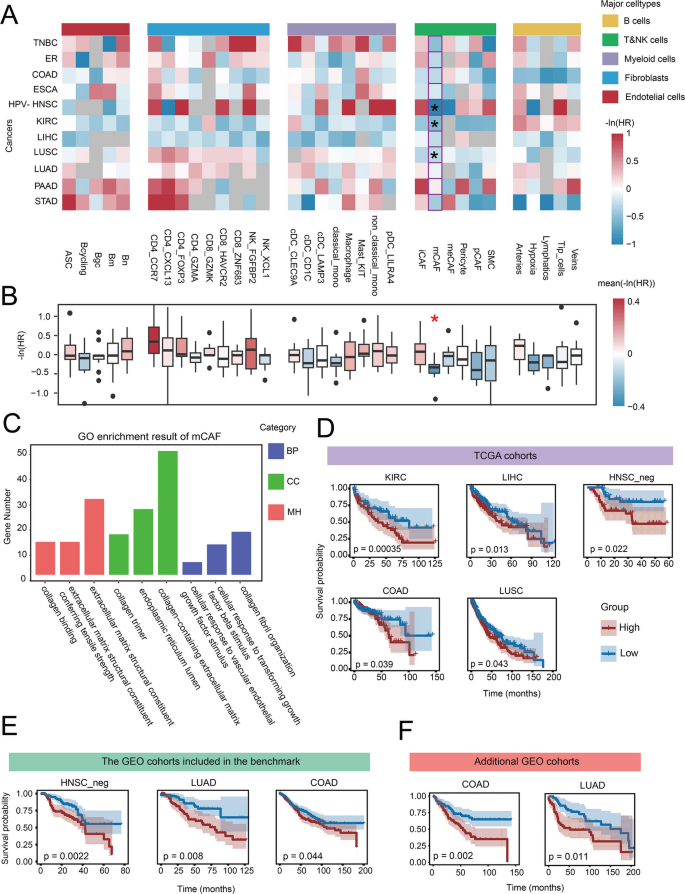
结果解读:ReCIDE方法识别到基质癌相关成纤维细胞(mCAF)为泛癌预后标志物(p=0.032),在ER+乳腺癌、HPV-头颈部鳞状细胞癌、肾透明细胞癌中与不良预后显著相关,在其他7种癌症中也呈现类似趋势;GO富集分析显示mCAF的标记基因富集于“细胞对TGF-beta刺激的应答”“细胞对VEGF刺激的应答”等通路,提示其可能通过调控肿瘤血管生成促进肿瘤进展;联合预后指标在5个TCGA队列中均显示出显著的预后关联,在5个GEO基准队列中的3个队列中显著,在额外的2个GEO队列中,LUAD队列的p值分别为0.002和0.011,证明其跨队列的可靠性。
产品关联:文献未提及具体实验产品,领域常规使用Seurat包构建细胞图谱,clusterProfiler包进行GO富集分析,survival、survminer包进行生存分析。
4. Biomarker研究及发现成果解析
本研究发现的Biomarker包括细胞亚型标志物(基质癌相关成纤维细胞,mCAF)和联合细胞比例指标(经典单核细胞比例+mCAF比例),通过多队列的系统验证,证明了其泛癌预后价值,为肿瘤患者的预后分层提供了新的工具。
Biomarker定位:涉及的Biomarker类型为细胞亚型(mCAF)和联合细胞比例指标;筛选与验证逻辑为:基于ReCIDE反卷积方法的泛癌队列分析,通过多变量Cox回归和Wilcoxon符号秩检验筛选出mCAF为泛癌预后标志物;随后构建经典单核细胞+mCAF的联合指标,依次在TCGA队列、GEO基准队列、额外GEO队列中进行验证,形成“泛癌筛选→内部验证→外部验证”的完整逻辑链条。
研究过程详述:Biomarker的来源为肿瘤组织bulk RNA-seq数据的反卷积结果,即通过ReCIDE方法从bulk数据中估算的细胞类型比例;验证方法包括多变量Cox回归分析(控制混杂因素)、Kaplan-Meier生存分析、Wilcoxon符号秩检验;特异性与敏感性数据:mCAF的泛癌预后关联经FDR校正后p=0.032;联合预后指标在TCGA的5个队列中均显示显著的预后关联,在GEO的5个基准队列中的3个队列中显著,在额外的2个GEO队列中,LUAD队列的p值分别为0.002和0.011;样本量方面,泛癌分析包含11种癌症实体的TCGA队列,验证队列包含5个GEO基准队列及2个额外GEO队列,总样本量超过5891例。
核心成果提炼:mCAF作为泛癌预后标志物,在多癌种中与不良预后相关,风险比(HR)的-ln(HR)值在多数癌症中<0,提示其可作为独立的预后预测因子;联合预后指标(经典单核细胞比例+mCAF比例)具有跨癌种的预后预测能力,能有效对患者进行风险分层,其创新性在于首次基于真实bulk数据的反卷积方法筛选出泛癌预后细胞亚型,并构建了联合细胞比例指标,验证了其跨队列的可靠性;统计学结果显示,mCAF的泛癌预后关联p=0.032,联合指标在多个队列中p<0.05,具有显著的统计学意义。