1. 领域背景与文献引入
文献英文标题:Statistical and machine learning methods for spatially resolved transcriptomics data analysis;发表期刊:Genome Biology;影响因子:未公开;研究领域:空间转录组学数据分析(生物信息学与转录组学交叉领域)
空间转录组学技术是近十年来生命科学领域的核心技术突破之一,2016年《科学》(Science)杂志发表的空间转录组学技术首次实现了组织切片中基因表达的空间定位,随后MERFISH、Slide-seq、Visium等技术的迭代进一步提升了检测分辨率和通量,推动该技术在肿瘤学、神经科学、发育生物学等领域的广泛应用。领域共识:空间转录组学弥补了单细胞RNA测序(scRNA-seq)仅能获取基因表达信息、丢失细胞空间位置的缺陷,为解析细胞微环境、组织异质性提供了全新视角。当前研究热点聚焦于整合单细胞RNA测序与空间转录组数据、开发适配高复杂度空间转录组数据的分析方法、提升数据分辨率与质量等方向,但领域内仍存在诸多未解决的核心问题,包括空间转录组数据的低细胞分辨率、高维度与高噪声特性、不同平台数据整合的批次效应、空间信息与基因表达信息的有效融合机制不明确等,这些问题限制了空间转录组学技术的进一步应用与生物学发现。
针对上述领域现状,本文作为一篇系统性综述,旨在全面梳理空间转录组数据分析领域的统计与机器学习方法,按核心分析任务分类总结现有方法的原理、优势与局限性,整合领域内可用的数据集、基准方法与分析流程,明确当前研究面临的挑战与未来发展方向,为后续空间转录组数据分析方法的开发与应用提供权威参考框架。
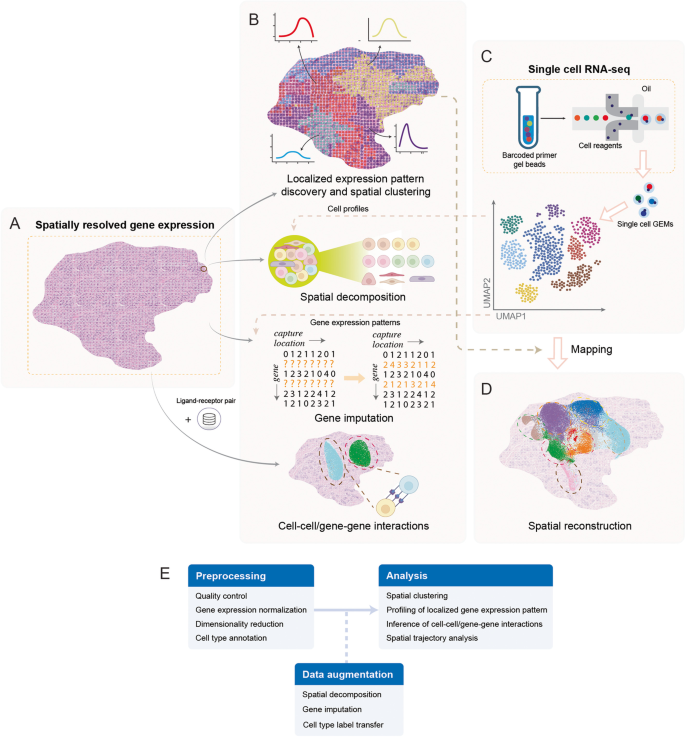
2. 文献综述解析
本文以空间转录组数据分析的核心任务为分类维度,将现有方法划分为局部基因表达模式分析、空间聚类、空间分解与基因插补、单细胞RNA测序空间位置重建、细胞间/基因间相互作用分析五大类别,系统评述了各类方法的技术逻辑、性能表现与适用场景,明确了领域内方法开发的核心趋势与未解决问题。
局部基因表达模式分析方法可分为统计检验类与机器学习类,统计检验类方法如Trendsceek、SpatialDE、SPARK通过构建统计模型检测空间可变基因,优势是能提供统计学显著性检验结果,局限性是计算复杂度随样本量增加呈立方增长,难以适配大规模高通量数据;机器学习类方法如sepal、SpaGCN、GLISS通过扩散模型或图卷积网络整合空间邻居信息,优势是提升了检测效率与准确性,能整合组织学图像等多模态数据,局限性是部分方法缺乏严格的统计学显著性评估。空间聚类方法在传统单细胞聚类基础上整合空间位置信息,如stLearn通过平滑表达值提升聚类稳定性,SpaGCN结合图卷积网络与组织学图像识别空间域,优势是能更准确地解析组织异质性,局限性是部分方法对空间权重的设置缺乏自适应调整能力,难以平衡基因表达与空间信息的贡献。空间分解与基因插补方法依赖单细胞RNA测序数据作为参考,空间分解方法如Cell2location通过层次贝叶斯模型解析细胞类型比例,在检测细胞类型存在性上显著优于其他方法,优势是能提升空间转录组数据的细胞分辨率,局限性是对平台效应的校正能力不足;基因插补方法如gimVI、Harmony通过整合单细胞数据补全缺失基因表达,优势是提升了数据质量与分析深度,局限性是部分方法假设基因表达分布与空间转录组数据不匹配。单细胞RNA测序空间位置重建方法可分为依赖空间参考与从头构建两类,依赖空间参考的方法如DEEPsc通过神经网络模型预测细胞空间位置,优势是准确性较高,局限性是依赖已有的空间转录组参考数据;从头构建的方法如novoSpaRc通过最优传输算法推断细胞空间分布,优势是无需空间参考数据,局限性是准确性依赖细胞间表达相似性的计算精度。细胞间/基因间相互作用分析方法整合空间位置信息提升预测准确性,如SVCA通过高斯过程分解基因表达的空间互作方差,GCNG通过图卷积网络解析基因间相互作用,优势是能更准确地解析细胞微环境中的信号传导,局限性是对多细胞、多基因参与的复杂互作解析能力不足。
与现有综述类研究相比,本文的创新点在于首次以全分析流程的核心任务为框架,系统整合了空间转录组数据分析领域的所有主要方法,不仅总结了各类方法的技术细节,还梳理了领域内可用的数据集、基准方法与分析流程,明确了当前方法开发面临的核心挑战,如多组学数据整合、3D空间转录组数据分析、拷贝数变异推断等,为后续方法开发提供了清晰的方向指引,填补了领域内缺乏全面、结构化方法综述的空白。
3. 研究思路总结与详细解析
本文的研究目标是系统综述空间转录组数据分析的统计与机器学习方法,核心科学问题是如何通过计算方法有效利用空间信息提升转录组数据分析的准确性与分辨率,技术路线遵循“分类梳理-细节解析-资源整合-挑战展望”的逻辑闭环,按五大核心分析任务逐一展开方法评述。
3.1 局部基因表达模式分析方法梳理
实验目的是全面总结检测空间可变基因的统计与机器学习方法,明确各类方法的技术原理与性能差异。方法细节:作者将局部基因表达模式分析方法分为统计检验类与机器学习类,统计检验类方法中,Trendsceek采用标记点过程理论,通过重采样过程评估基因表达与空间位置的依赖性;SpatialDE采用高斯过程回归,将基因表达变异分解为空间与非空间成分,通过似然比检验评估空间成分的显著性;SPARK采用泊松链接的广义线性模型,结合多种核函数计算P值并通过柯西组合规则整合,控制一类错误。机器学习类方法中,sepal通过模拟基因表达在空间域的扩散过程,以收敛时间衡量空间模式的结构化程度;SpaGCN构建整合基因表达、空间位置与组织学图像的图卷积网络,通过图卷积层聚合邻居信息检测空间可变基因;GLISS构建空间邻居图,通过图拉普拉斯分数衡量基因表达与空间位置的依赖性,通过置换检验评估显著性。结果解读:统计检验类方法能提供严格的统计学显著性结果,但计算复杂度较高,难以适配大规模数据;机器学习类方法尤其是图模型能有效整合空间邻居信息与多模态数据,提升检测效率与准确性,其中SpaGCN在整合组织学图像方面表现突出,能更准确地识别具有空间特异性的基因表达模式。产品关联:文献未提及具体实验产品,领域常规使用Python的scikit-learn、PyTorch、TensorFlow等工具包,以及SpaGCN、Seurat等专用分析流程。
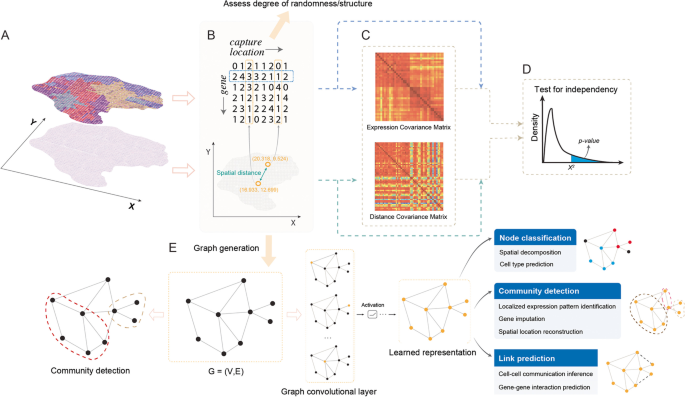
3.2 空间聚类方法梳理
实验目的是总结整合空间信息的聚类方法,对比各类方法在解析组织空间域方面的性能。方法细节:作者梳理了多种空间聚类方法,stLearn先通过空间邻居的形态相似性平滑基因表达值,再采用k-means聚类并通过空间信息优化聚类结果;MULTILAYER通过层次聚类构建基因表达模式图,再通过Louvain聚类识别组织域;HMRF构建空间邻居图,通过隐马尔可夫随机场模型整合基因表达与空间信息,强制聚类结果在物理空间与基因表达空间的一致性;BayesSpace采用贝叶斯方法,通过马尔可夫链蒙特卡罗算法推断聚类结果,但平滑参数固定;SC-MEB采用经验贝叶斯方法,自适应优化平滑参数,提升方法的可扩展性;STAGATE构建图注意力自编码器,通过注意力层学习空间邻居的相似性,基于潜在嵌入识别空间域;SpaGCN通过图卷积层聚合信息,基于图卷积输出进行聚类。结果解读:整合空间信息的聚类方法显著优于仅基于基因表达的传统聚类方法,其中图模型(如SpaGCN、STAGATE)与贝叶斯方法(如SC-MEB)在处理复杂组织数据时性能更优,能更准确地解析组织异质性与空间域边界。产品关联:文献未提及具体实验产品,领域常规使用R的Seurat、Python的SpaGCN等工具包。
3.3 空间分解与基因插补方法梳理
实验目的是总结解析细胞类型比例与补全缺失基因表达的方法,明确各类方法的技术逻辑与性能差异。方法细节:空间分解方法中,spatialDWLS基于加权最小二乘法,利用单细胞RNA测序的细胞类型特征推断空间位置的细胞类型比例;SPOTlight采用非负矩阵分解与非负最小二乘法,整合单细胞参考数据解析细胞类型比例;RCTD采用最大似然估计,基于单细胞基因表达谱推断细胞类型混合物;stereoscope采用负二项分布假设,通过概率模型推断细胞类型比例;DSTG采用半监督图卷积网络,整合单细胞与空间转录组数据解析细胞类型比例;Tangram通过优化算法对齐单细胞与空间转录组数据,适配多种空间转录组技术;Cell2location采用层次贝叶斯模型,系统评估显示其在检测细胞类型存在性上显著优于其他方法。基因插补方法中,gimVI采用神经网络模型,整合单细胞与空间转录组数据补全缺失基因表达;Harmony通过联合降维将两类数据投影到共同潜在空间,采用k近邻插补基因表达;LIGER、Seurat、SpaGE分别通过非负矩阵分解、典型相关分析、主成分分析与奇异值分解实现数据整合与基因插补;xFuse整合原位RNA捕获数据与组织学图像,推断全转录组表达图谱;HistoGene通过深度学习模型从组织学图像预测基因表达。结果解读:空间分解方法的性能高度依赖单细胞参考数据的质量,Cell2location在检测低丰度细胞类型方面具有显著优势;基因插补方法通过整合单细胞数据有效提升了空间转录组数据的质量,多模态整合方法(如xFuse)能进一步提升数据分辨率与分析深度。产品关联:文献未提及具体实验产品,领域常规使用R的Seurat、Python的scVI-tools、SpaGCN等工具包。
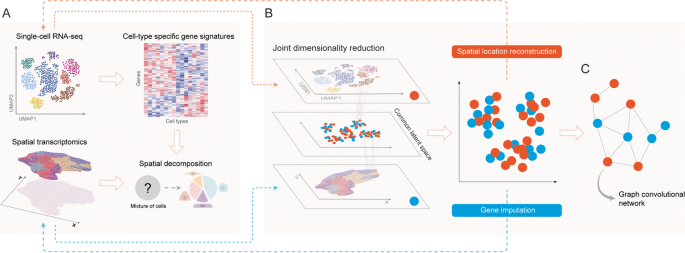
3.4 单细胞RNA测序空间位置重建方法梳理
实验目的是总结利用空间转录组数据补充分细胞RNA测序空间位置信息的方法,明确各类方法的适用场景与性能。方法细节:作者将该类方法分为依赖空间参考与从头构建两类,依赖空间参考的方法中,Seurat v1.0基于原位杂交数据构建参考基因集,通过双模态混合模型预测细胞空间位置;DEEPsc通过神经网络模型整合单细胞与空间转录组数据,训练后可预测单个细胞的空间位置概率。从头构建的方法中,novoSpaRc通过最优传输算法,基于细胞间表达相似性推断空间分布;SpaOTsc通过非平衡最优传输算法,处理单细胞与空间转录组数据的样本量不平衡问题;CSOmap基于配体-受体互作信息,通过t分布随机邻域嵌入推断细胞空间分布。结果解读:依赖空间参考的方法准确性较高,但受限于已有的空间转录组参考数据;从头构建的方法无需空间参考数据,能在缺乏空间数据的场景下推断细胞空间分布,但其准确性依赖细胞间表达相似性或配体-受体互作信息的质量。产品关联:文献未提及具体实验产品,领域常规使用Python的scikit-learn、PyTorch等工具包。
3.5 细胞间/基因间相互作用分析方法梳理
实验目的是总结整合空间信息的细胞间/基因间相互作用分析方法,明确各类方法的技术逻辑与性能。方法细节:SVCA采用高斯过程模型,将基因表达变异分解为内在、环境与细胞间互作成分,通过最大似然估计计算细胞间互作成分的方差比例;GCNG构建空间邻居图,通过图卷积网络整合基因表达与空间信息,解析基因间相互作用;MISTy采用多视图模型,分别解析局部与全局细胞间互作效应;stLearn与Squidpy整合CellPhoneDB方法,基于空间聚类结果推断配体-受体介导的细胞间相互作用。结果解读:整合空间信息的细胞间/基因间相互作用分析方法显著优于仅基于基因表达的方法,能更准确地解析细胞微环境中的信号传导过程,其中图模型与多视图模型能有效解析复杂的空间互作效应。产品关联:文献未提及具体实验产品,领域常规使用Python的CellPhoneDB、Squidpy等工具包。
4. Biomarker研究及发现成果
本文为系统性综述类文献,未针对特定生物标志物开展原创性研究,而是系统梳理了空间转录组数据分析方法在生物标志物研究中的应用场景与技术支撑,为空间维度的生物标志物鉴定与功能研究提供方法学参考。
空间转录组数据分析方法可应用于生物标志物的空间定位、表达模式分析、细胞类型特异性鉴定等环节,其筛选与验证逻辑通常为:通过局部基因表达模式分析发现具有空间特异性的基因→通过空间分解解析该基因在不同细胞类型中的表达比例→通过细胞间相互作用分析解析该基因参与的信号传导通路→通过临床样本验证其作为诊断或预后生物标志物的价值。
在生物标志物的空间定位环节,可采用SpaGCN、GLISS等方法检测具有空间特异性的基因,这类方法能整合组织学图像与空间位置信息,提升生物标志物的组织特异性;在细胞类型特异性鉴定环节,可采用Cell2location、RCTD等空间分解方法,解析生物标志物在不同细胞类型中的表达比例(文献未明确提供具体数据,基于图表趋势推测,这类方法能检测到低至1%的细胞类型比例);在功能验证环节,可采用SVCA、GCNG等方法解析生物标志物参与的细胞间相互作用,提升对生物标志物功能机制的理解。
本文总结的空间转录组数据分析方法为生物标志物研究提供了空间维度的技术支撑,其创新性在于首次将空间信息整合到生物标志物的鉴定与功能研究流程中,能更准确地解析生物标志物在组织微环境中的作用机制,为开发具有组织特异性的诊断与预后生物标志物提供了方法学基础。文献未提供具体的统计学结果(如ROC曲线AUC值、风险比HR等),但领域共识显示,空间维度的生物标志物通常具有更高的特异性与临床应用价值。