1. 领域背景与文献引入
文献英文标题:Retinoic acid has different effects on UCP1 expression in mouse and human adipocytes;发表期刊:BMC Cell Biology;影响因子:未公开;研究领域:脂肪细胞代谢与肥胖治疗。
哺乳动物脂肪组织分为白色脂肪组织(WAT)和棕色脂肪组织(BAT),前者负责能量储存,后者通过解偶联蛋白1(UCP1)介导的产热作用消耗能量,是肥胖防治的潜在靶点。领域共识:激活棕色脂肪或诱导白色脂肪棕色化(即产生米色脂肪)已成为肥胖及代谢综合征治疗的前沿策略。视黄酸(RA,维生素A的活性形式)作为核受体激动剂,已被证实可在小鼠脂肪细胞中显著诱导解偶联蛋白1表达,促进产热;但人类脂肪细胞对视黄酸的响应性研究较为零散,缺乏系统的物种间对比数据,这一空白限制了视黄酸在人类肥胖治疗中的转化应用。因此,本研究旨在系统对比全反式视黄酸(ATRA)对小鼠和人类多种脂肪细胞模型中解偶联蛋白1表达的调控作用,并解析其分子机制,为视黄酸的临床应用提供实验依据。
2. 文献综述解析
作者对现有研究的分类维度主要为物种差异(小鼠与人类)、作用通路(核受体介导的转录调控)及实验模型(细胞系与原代细胞)。现有研究的关键结论包括:在小鼠模型中,视黄酸通过激活视黄酸受体(RAR)上调解偶联蛋白1表达,促进棕色脂肪产热及白色脂肪棕色化,且高剂量视黄酸可抑制脂肪细胞分化;部分初步研究显示人类脂肪细胞对视黄酸的响应与小鼠存在差异,但未形成系统结论。技术方法优势方面,既往研究利用小鼠细胞系及体内实验明确了视黄酸调控解偶联蛋白1的核心通路,为机制研究奠定了基础;但局限性也较为明显,包括缺乏直接对比小鼠与人类脂肪细胞的系统实验,未覆盖多种人类脂肪细胞模型(如原代细胞),且对视黄酸作用的受体亚型、是否依赖过氧化物酶体增殖物激活受体γ共激活因子1α(PGC-1α)等关键问题尚未明确。本研究的创新价值在于,首次系统整合了小鼠和人类的多种脂肪细胞模型(细胞系、原代细胞),通过慢性与急性处理实验明确了视黄酸调控解偶联蛋白1的物种特异性,同时解析了视黄酸作用的受体亚型,并排除了过氧化物酶体增殖物激活受体δ(PPARδ)及PGC-1α的介导作用,填补了物种间调控差异的研究空白,为视黄酸在肥胖治疗中的转化应用提供了关键实验证据。
3. 研究思路总结与详细解析
本研究的核心目标是明确全反式视黄酸(ATRA)对小鼠与人类脂肪细胞解偶联蛋白1表达的调控差异及分子机制;核心科学问题为视黄酸调控解偶联蛋白1的物种特异性分子基础;技术路线遵循“模型构建→处理干预→分子检测→机制验证→跨物种验证”的闭环逻辑,通过多组平行实验系统对比物种间的响应差异,并解析关键调控通路。
3.1 小鼠脂肪细胞分化期ATRA慢性处理实验
实验目的:探究ATRA对分化中小鼠脂肪细胞解偶联蛋白1表达及脂肪分化的影响;方法细节:选取4种小鼠脂肪细胞模型(3T3-L1白色前脂肪细胞、野生型小鼠胚胎成纤维细胞(WT MEF)、C3H10T½间充质干细胞、视网膜母细胞瘤基因敲除(Rb-/-)MEF棕色脂肪模型),在细胞汇合前2天至分化第8天期间,给予10nM至10μM梯度浓度的ATRA处理,第8天通过逆转录定量聚合酶链反应(RT-qPCR)检测脂肪分化标记基因脂肪酸结合蛋白4(FABP4)、解偶联蛋白1及RARβ的mRNA水平,通过免疫印迹检测解偶联蛋白1蛋白水平;结果解读:RARβ表达随ATRA剂量增加呈剂量依赖性升高(Rb-/- MEF除外),FABP4表达在高剂量ATRA(10μM)时显著降低,提示脂肪分化被抑制;中剂量ATRA(3T3-L1为1μM,WT MEF为0.1μM和1μM,C3H10T½和Rb-/- MEF为0.1μM)可显著诱导解偶联蛋白1的mRNA和蛋白表达,其中WT MEF来源脂肪细胞中解偶联蛋白1 mRNA上调18倍,3T3-L1上调4倍,C3H10T½上调4倍,Rb-/- MEF上调3倍(n=3,P<0.05);产品关联:文献未提及具体实验产品,领域常规使用RT-qPCR试剂盒、免疫印迹(WB)相关抗体及蛋白电泳系统等。
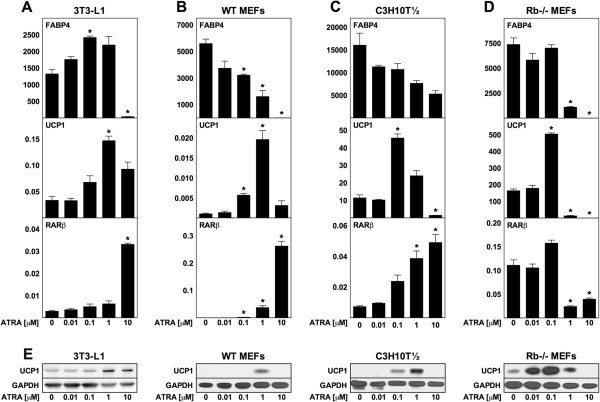
3.2 成熟小鼠脂肪细胞ATRA急性处理实验
实验目的:探究ATRA对成熟小鼠脂肪细胞解偶联蛋白1表达的影响;方法细节:在小鼠脂肪细胞分化第8天(成熟阶段),给予1μM ATRA处理24小时,第9天检测FABP4、解偶联蛋白1及RARβ的mRNA和蛋白水平;结果解读:WT MEF、Rb-/- MEF及C3H10T½来源的成熟脂肪细胞中,解偶联蛋白1 mRNA分别上调15倍、4.6倍和2.5倍(n=3,P<0.05),蛋白水平与mRNA表达趋势一致;而3T3-L1成熟脂肪细胞中解偶联蛋白1表达无显著变化;FABP4 mRNA水平在MEF来源脂肪细胞中略有降低,在3T3-L1和C3H10T½中无显著变化;产品关联:文献未提及具体实验产品,领域常规使用RT-qPCR试剂盒、免疫印迹相关抗体等。
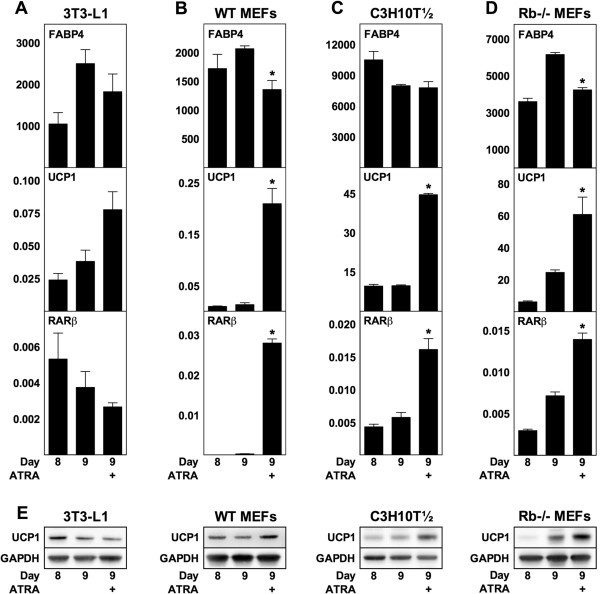
3.3 RAR介导ATRA作用的特异性验证实验
实验目的:明确ATRA调控解偶联蛋白1表达的受体机制;方法细节:首先用泛RAR激动剂TTNPB(不激活PPARδ)对WT MEF进行慢性(分化期)和急性(成熟阶段)处理,检测相关基因表达;随后用泛RAR拮抗剂BMS493与ATRA共处理,验证RAR的介导作用;最后用RAR亚型选择性激动剂(AM580为RARα选择性,tazarotene为RARβ/γ选择性,CD1530为RARγ选择性)处理WT MEF,检测解偶联蛋白1表达;结果解读:TTNPB可模拟ATRA的作用,慢性处理时最高诱导解偶联蛋白1 mRNA上调25倍,急性处理时上调6倍(n=3,P<0.05);BMS493可消除慢性处理中ATRA对解偶联蛋白1的诱导作用,并减弱急性处理中的诱导效果;三种RAR亚型激动剂均能显著诱导解偶联蛋白1表达,其中AM580和tazarotene的诱导作用最强,提示任意RAR亚型激活均可介导ATRA的作用;产品关联:文献未提及具体实验产品,领域常规使用核受体激动剂/拮抗剂试剂、RT-qPCR试剂盒等。
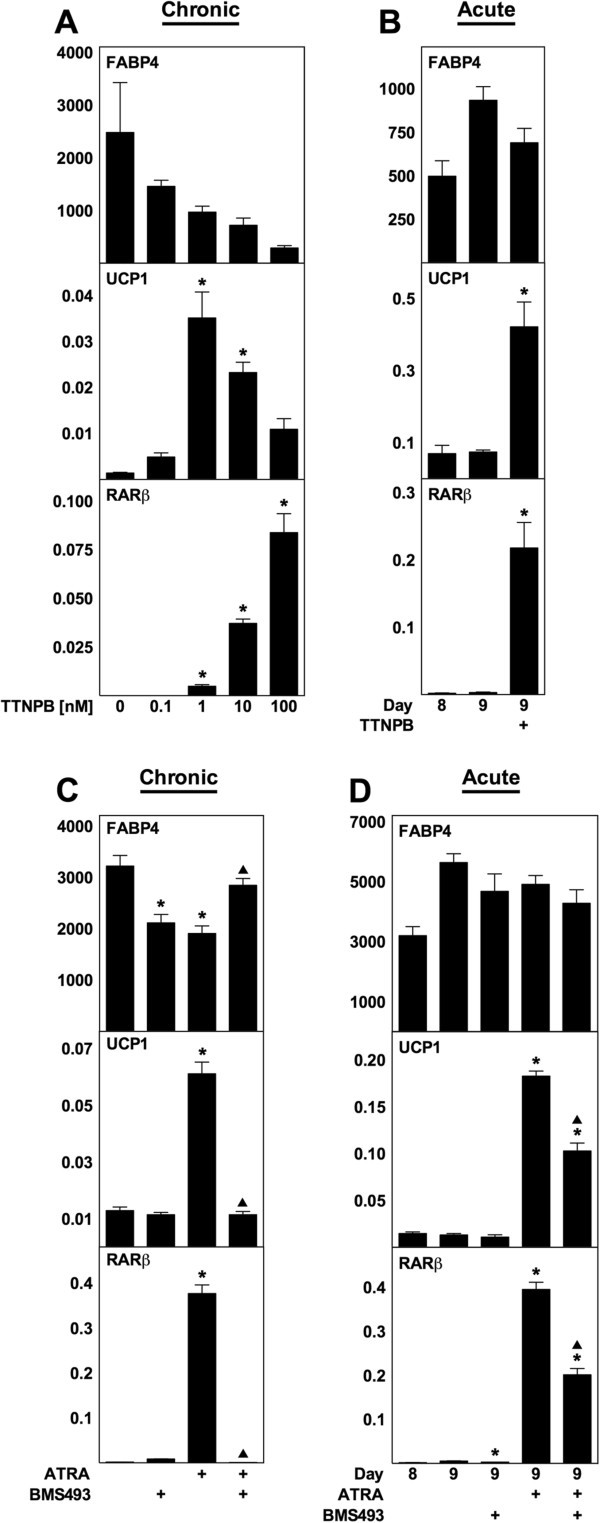
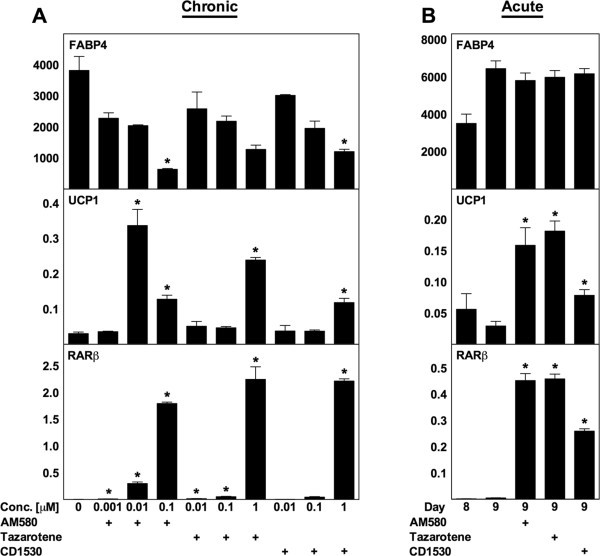
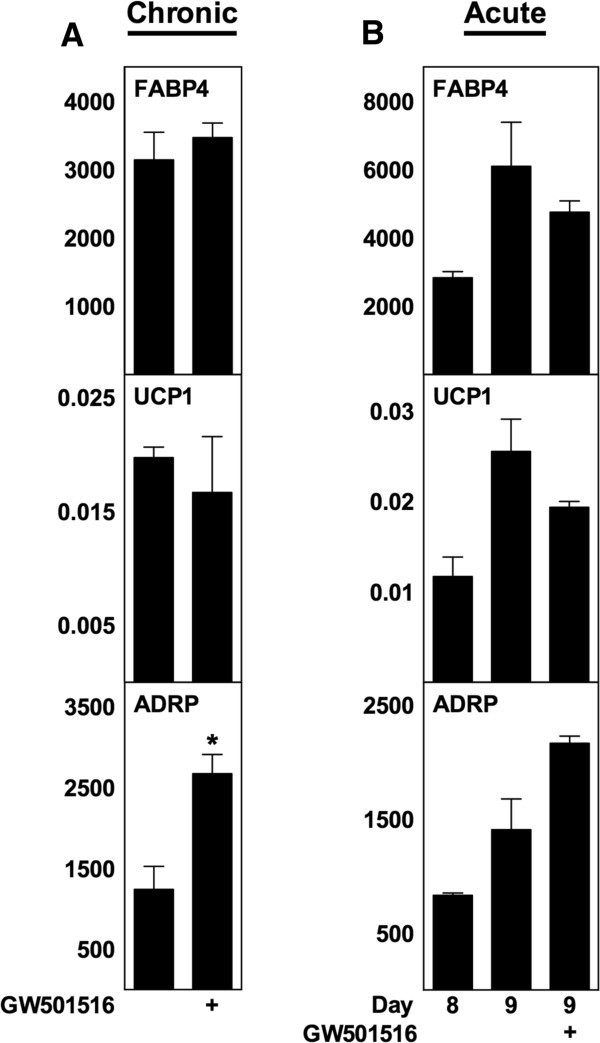
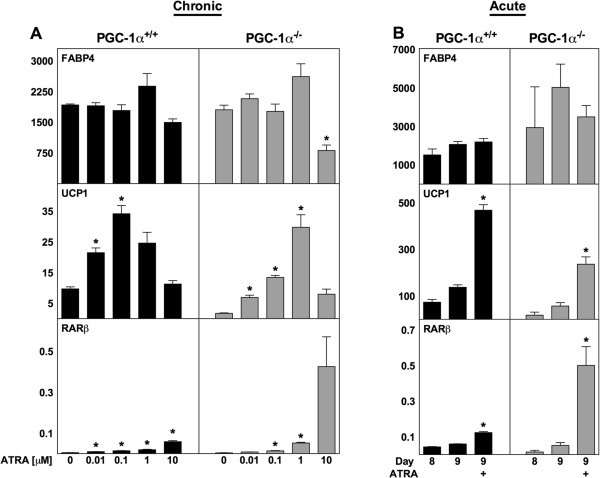
3.4 PPARδ与PGC-1α的作用排除实验
实验目的:验证PPARδ和PGC-1α是否参与ATRA对解偶联蛋白1的调控;方法细节:用PPARδ激动剂GW501516对WT MEF进行慢性和急性处理,检测解偶联蛋白1及PPARδ靶基因脂肪分化相关蛋白(ADRP)的表达;同时利用PGC-1α+/+和PGC-1α-/-的永生化棕色前脂肪细胞系,给予ATRA慢性和急性处理,检测解偶联蛋白1表达;结果解读:GW501516可显著上调ADRP表达(n=3,P<0.05),但对解偶联蛋白1表达无显著影响;PGC-1α-/-棕色脂肪细胞中,ATRA仍能显著诱导解偶联蛋白1表达,其中1μM ATRA处理时mRNA上调17倍(n=3,P<0.05),提示ATRA的作用不依赖PGC-1α;产品关联:文献未提及具体实验产品,领域常规使用核受体激动剂、基因敲除细胞系、RT-qPCR试剂盒等。
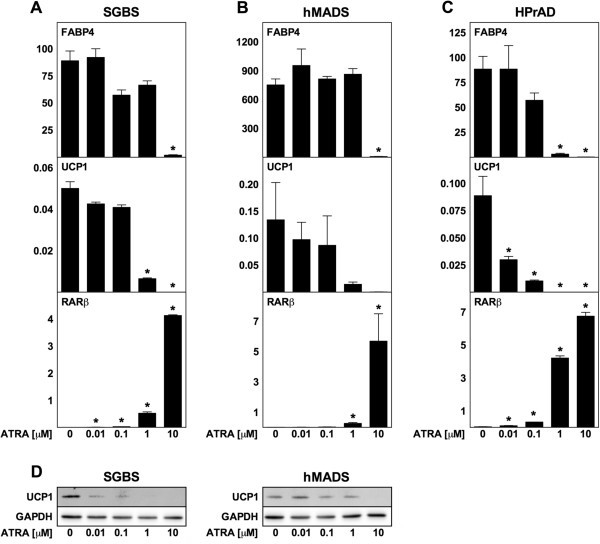
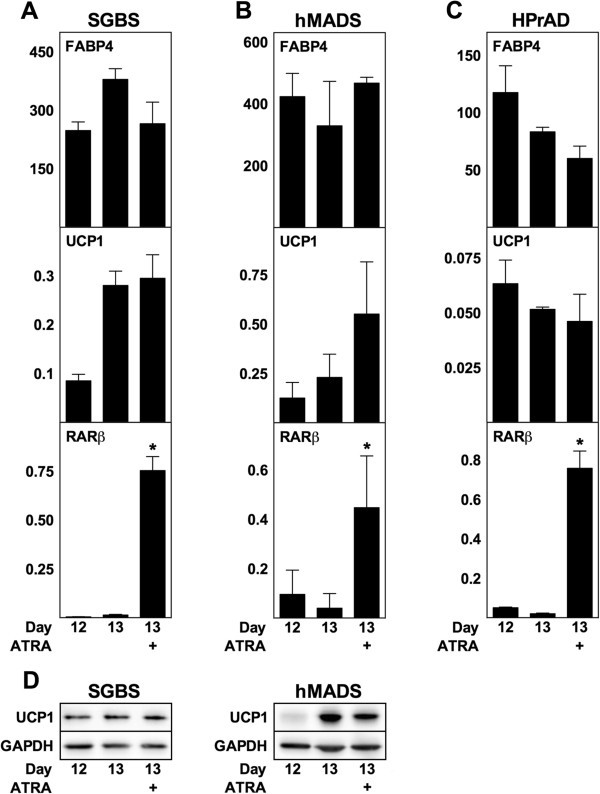
3.5 人类脂肪细胞ATRA处理实验
实验目的:探究ATRA对人类脂肪细胞解偶联蛋白1表达及分化的影响;方法细节:选取3种人类脂肪细胞模型(SGBS白色前脂肪细胞、hMADS多能脂肪干细胞、原代人类白色前脂肪细胞),进行慢性(分化期-2天至12天)和急性(成熟阶段第12天处理24小时)ATRA处理,检测FABP4、解偶联蛋白1及RARβ的mRNA和蛋白水平;结果解读:高剂量ATRA(10μM)可抑制SGBS、hMADS及原代细胞的脂肪分化(FABP4降低,n=3,P<0.05),RARβ表达随ATRA剂量增加呈剂量依赖性升高;但所有浓度的ATRA均未诱导解偶联蛋白1的mRNA或蛋白表达,甚至在原代细胞中10nM ATRA即可显著抑制解偶联蛋白1的基础表达;急性处理也未引起解偶联蛋白1表达的显著变化;产品关联:文献未提及具体实验产品,领域常规使用人类脂肪细胞培养试剂、RT-qPCR试剂盒、免疫印迹相关抗体等。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究中涉及的生物标志物为解偶联蛋白1(UCP1),属于功能型生物标志物,用于表征脂肪细胞的产热活性;其筛选与验证逻辑为:基于小鼠模型中ATRA诱导解偶联蛋白1的已知结论,在人类脂肪细胞模型中系统验证其响应性,明确物种间的调控差异,验证链条覆盖“细胞系→原代细胞”“慢性处理→急性处理”多个维度。
研究过程详述
解偶联蛋白1的来源为小鼠和人类的脂肪细胞(包括细胞系及原代细胞);验证方法包括RT-qPCR定量检测mRNA水平、免疫印迹检测蛋白水平;特异性与敏感性数据显示,在小鼠脂肪细胞中,中剂量ATRA(0.1μM-1μM)可特异性诱导解偶联蛋白1表达,mRNA上调倍数最高达18倍(n=3,P<0.05);而在人类脂肪细胞中,无论慢性或急性处理、低剂量或高剂量ATRA,均无法诱导解偶联蛋白1表达,甚至在原代细胞中低剂量ATRA即可抑制其基础表达,显示出显著的物种特异性。
核心成果提炼
本研究的核心成果是明确了解偶联蛋白1作为产热生物标志物,其对ATRA的响应存在严格的物种特异性:在小鼠脂肪细胞中,ATRA通过激活任意RAR亚型上调解偶联蛋白1表达,该过程不依赖PPARδ及PGC-1α;而在人类脂肪细胞中,ATRA无法诱导解偶联蛋白1表达,甚至抑制其基础水平。该成果的创新性在于首次系统对比了小鼠与人类多种脂肪细胞模型的响应差异,填补了物种间调控机制的研究空白;统计学结果显示所有实验均重复3次(n=3),差异显著组P<0.05。这一发现为肥胖治疗中基于解偶联蛋白1的策略提供了重要参考,提示小鼠模型的结论不能直接推广至人类,需开展更多人类特异性的产热调控研究。