1. 领域背景与文献引入
文献英文标题:Transcriptional regulatory network reveals key transcription factors for regulating agronomic traits in soybean;发表期刊:Genome Biology;影响因子:17.906(2023年);研究领域:作物分子生物学/大豆功能基因组学
植物转录因子(TF)与顺式调控元件(CRE)的互作是基因时空表达调控的核心,也是植物适应环境、产生表型多样性的分子基础。近年来,随着DAP-seq、ChIP-seq等技术的发展,动植物中已开展大规模TF结合图谱的研究,揭示了TF结合的基因组特征及调控规律。大豆作为全球最重要的油料作物之一,其农艺性状(如种子含油量、种皮颜色、耐旱性等)的遗传改良是育种的核心目标。虽然已有研究鉴定了少数调控大豆农艺性状的TF(如GmWRI1b调控含油量、GmE1调控开花期),但这些研究多聚焦单个TF或通路,缺乏全基因组尺度的TF-靶标互作网络,无法系统解析复杂农艺性状的调控层级,也限制了QTL区间内候选调控因子的精准定位。因此,构建大豆全基因组转录调控网络,系统挖掘农艺性状的关键调控TF,对加速大豆分子育种具有重要的学术价值和应用前景。
2. 文献综述解析
作者从TF-顺式元件互作的调控机制出发,梳理了动植物转录调控网络研究的技术进展与核心结论:在动物和模式植物中,DAP-seq、ChIP-seq等技术已被用于绘制大规模TF结合图谱,揭示了TF结合位点的基因组分布特征、与表观修饰的关系,以及TF结合差异对基因表达分化的影响;同时,整合多组学数据构建的调控网络已成为挖掘复杂性状关键调控因子的重要工具。针对大豆领域,作者总结了现有研究的局限性:已报道的大豆TF结合数据仅覆盖少数家族,缺乏全基因组尺度的TF结合图谱;现有研究多聚焦单个农艺性状的通路解析,未构建整合多组学数据的全局调控网络;QTL区间内的候选基因筛选仍受连锁不平衡的限制,无法精准定位关键调控TF。
通过对比领域内未解决的核心问题,本研究的创新价值凸显:首次通过DAP-seq技术绘制148个大豆TF的全基因组结合图谱,解析了大豆TF结合的基因组特征及与全基因组复制(WGD)旁系同源基因表达分化的关系;整合多组学数据构建了覆盖91%大豆TF的高置信度转录调控网络SoyGRN,包含244万条TF-靶标互作关系;利用SoyGRN系统挖掘了调控种子种皮颜色、含油量、耐旱性的关键TF,并通过功能验证确认了其调控作用;开发了在线平台SoyTFBase,为大豆研究社区提供了便捷的调控网络查询工具。
3. 研究思路总结与详细解析
本研究的核心目标是构建大豆全基因组转录调控网络,解析TF结合的基因组特征,挖掘调控农艺性状的关键TF;核心科学问题包括大豆TF结合规律对基因表达及农艺性状的调控机制,以及如何利用调控网络精准定位QTL中的候选调控因子;技术路线形成“实验图谱绘制→特征解析→网络构建→功能挖掘→平台开发”的完整闭环:首先通过DAP-seq绘制TF结合图谱,分析其基因组特征及与WGD旁系同源基因的关系;然后整合多组学数据构建SoyGRN并验证其可靠性;接着利用SoyGRN挖掘调控种皮颜色、含油量的关键TF并进行功能验证;最后利用SoyGRN定位QTL中的候选TF,并开发在线平台供研究使用。
3.1 大豆TF的DAP-seq图谱绘制与质量控制
实验目的是获取大豆TF的全基因组结合位点,验证DAP-seq数据的可靠性。研究克隆了230个参与发育、胁迫响应、养分利用的大豆TF,通过DAP-seq技术绘制全基因组结合图谱,以峰中读数比例(FRiP)>2%为标准筛选得到148个TF(来自28个家族);对11个随机选择的TF进行生物学重复实验,验证数据重复性;同时将GmbZIP67的DAP-seq数据与已发表的ChIP-seq数据对比,验证技术可靠性。结果显示,共鉴定3,041,762个TF结合峰,每个TF的结合峰中位数为5737个;9个TF的生物学重复样本Pearson相关系数>0.9,不可复现发现率(IDR)<0.05,证明数据重复性高;GmbZIP67的DAP-seq与ChIP-seq峰重叠显著(超几何检验,P=0),且结合基序高度相似,进一步验证了DAP-seq数据的可靠性。
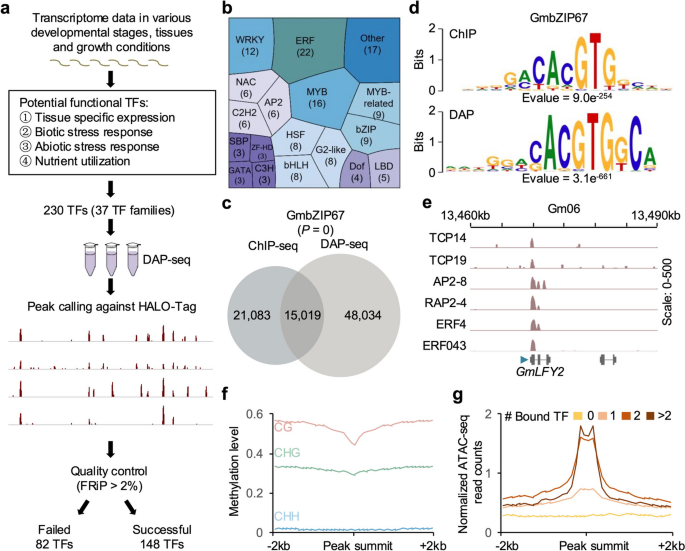
文献未提及具体实验产品,领域常规使用DAP-seq相关试剂盒(如NEBNext Ultra™ II DNA Library Prep Kit)、Illumina NovaSeq测序平台等。
3.2 大豆TF结合位点的基因组特征分析
实验目的是解析TF结合位点在基因组中的分布规律及与基因表达、表观修饰、遗传变异的关系。研究将所有TF的结合峰合并为2kb非重叠窗口进行主成分分析,分析TF结合位点在基因区域的分布、与染色质可及性(ATAC-seq)、DNA甲基化(MethylC-seq)的关系,并基于302份大豆重测序数据分析TF结合位点的遗传变异特征。结果显示,TF结合位点并非随机分布,而是富集于染色质开放区域(OCRs),形成TF结合热点区域(HOT区域,结合≥74个TF),这些区域富含二价组蛋白标记(H3K4me3和H3K27me3);启动子区结合TF数量越多的基因,表达水平越高(转录本每百万读数,TPM值更高),组织特异性越低(表达变异系数CV更小);TF结合位点的核苷酸多样性显著高于侧翼区域,且稀有SNP密度更高,说明TF结合位点是大豆基因组的遗传变异热点。
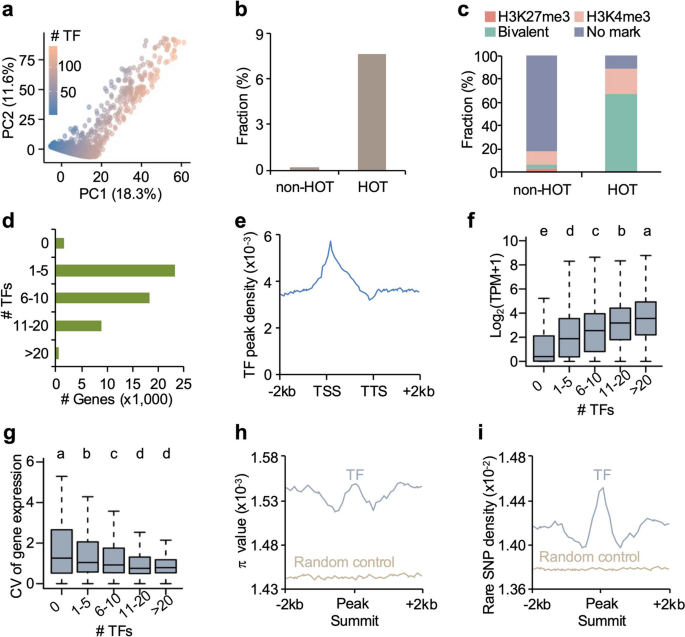
文献未提及具体实验产品,领域常规使用ATAC-seq、MethylC-seq技术及deepTools、MACS2等分析工具。
3.3 全基因组复制旁系同源基因的TF结合差异分析
实验目的是探究WGD旁系同源基因的TF结合差异对其表达分化的影响。研究在大豆基因组中鉴定了16,634对WGD旁系同源基因,分析其启动子区TF结合的重叠比例,对比TF结合差异与表达分化的相关性,并解析TF结合差异的分子机制。结果显示,89.6%的WGD旁系同源基因启动子区结合的TF重叠比例<50%,且TF结合差异与表达分化呈正相关(Wilcoxon秩和检验,P<0.01);99.3%的旁系同源基因具有相同的TF结合基序,但结合TF的位点甲基化水平显著低于未结合的位点(Wilcoxon配对秩和检验,P<0.01),说明DNA甲基化差异是导致WGD旁系同源基因TF结合差异的重要原因。
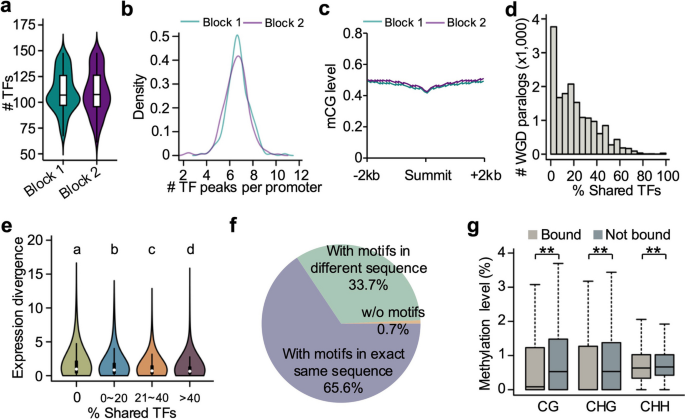
文献未提及具体实验产品,领域常规使用PlantTFDB等基因组注释工具及GATK、VCFtools等重测序数据分析工具。
3.4 大豆转录调控网络SoyGRN的构建与验证
实验目的是整合多组学数据构建高置信度的大豆TF-靶标互作网络,并验证其可靠性。研究整合了7个独立的调控网络:DAP-seq直接鉴定的TF-靶标互作(SoyDAP)、基因共表达网络(GENIE3、PCC)、进化保守调控网络(cCOE)、拟南芥DAP-seq同源映射(AtDAP)、基序扫描(PWM)、染色质可及性辅助的互作(OCR),通过严格过滤得到高置信度互作关系;同时利用已发表的GmMYB14过表达RNA-seq数据、ABI3-1的ChIP-seq数据验证网络准确性,并对网络进行模块化功能分析。结果显示,构建的SoyGRN包含244万条TF-靶标互作关系,涉及3188个TF和51665个靶基因,覆盖91%的大豆TF;GmMYB14的预测靶基因与过表达株系的差异表达基因显著重叠(超几何检验,P=2.7e-16),ABI3-1的预测靶基因与ChIP-seq鉴定的靶基因显著重叠(超几何检验,P=9.6e-55),证明网络准确性高;网络被划分为9个功能模块,每个模块富集特定的生物学过程,如M4模块富集蛋白质代谢过程,M7模块富集光合作用过程。
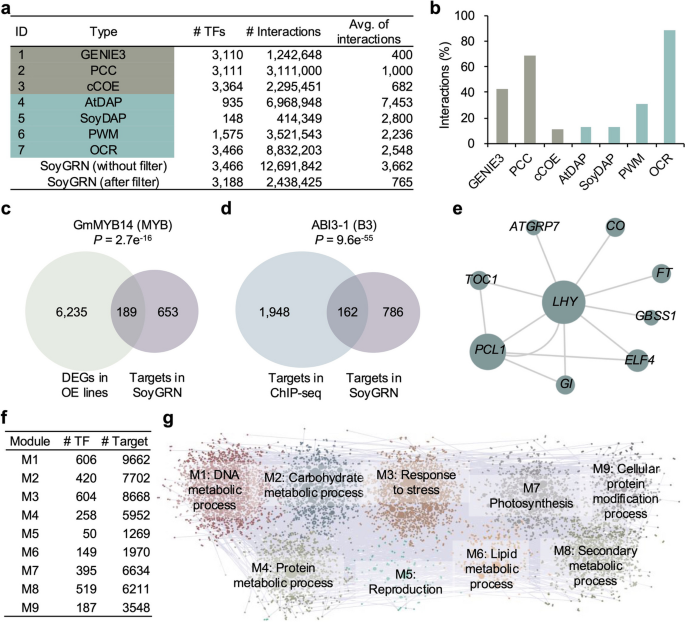
文献未提及具体实验产品,领域常规使用GENIE3、Cytoscape等网络构建与可视化工具。
3.5 调控种子种皮颜色的关键TF鉴定与功能验证
实验目的是利用SoyGRN挖掘调控大豆种皮颜色的关键TF,并验证其功能。研究收集了3179个参与类黄酮和花青素生物合成的基因,通过超几何检验筛选与这些基因互作的TF;对候选TF进行系统发育分析,选择与拟南芥TT8同源的GmTT8b进行过表达功能验证;同时分析GmTT8b及其靶基因的组织表达模式和染色质可及性。结果显示,共筛选到79个调控种皮颜色的候选TF,其中25个属于MYB家族;GmTT8b过表达株系的种子种皮颜色显著加深,且GmTT8b及其7个靶基因在种子中优先表达,其靶基因启动子区在种子中的染色质可及性更高,证明GmTT8b是调控大豆种皮颜色的关键TF。
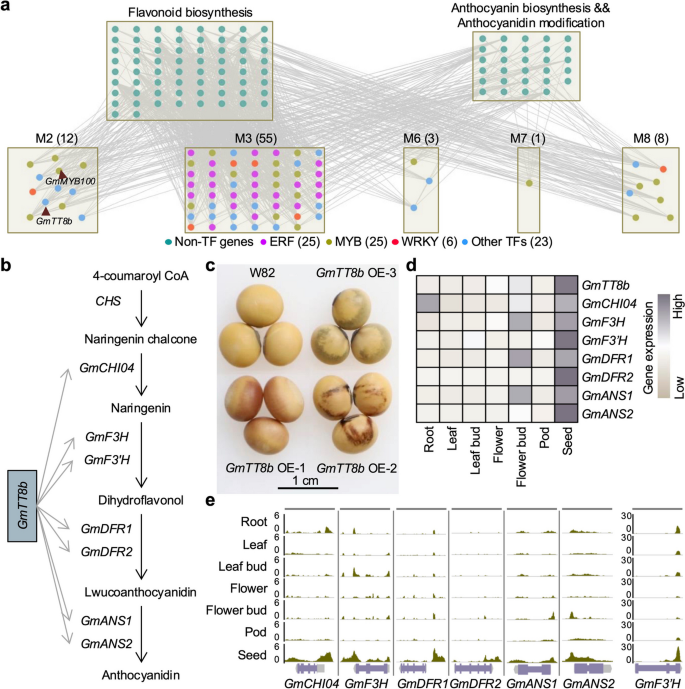
实验所用关键产品:CRISPR/Cas9载体(pYLCRISPR/Cas9P35S-BS)、农杆菌EHA101/EHA105、PrimeScript RT Reagent Kit反转录试剂盒、SYBR Green Master Mix荧光定量试剂。
3.6 调控种子含油量的关键TF鉴定与功能验证
实验目的是利用SoyGRN挖掘调控大豆种子含油量的关键TF,并验证其功能。研究收集了723个参与油脂积累的基因(包括脂肪酸合成、三酰甘油合成等通路),筛选与这些基因互作的TF;分析候选TF在种子发育不同阶段的表达模式;利用已构建的大豆突变体库筛选含油量变化的TF突变体,对SBP家族TF进行CRISPR/Cas9敲除验证。结果显示,共筛选到279个调控含油量的候选TF,包括已知的GmWRI1b、GmZF351等;SBP家族TF调控21.4%的脂肪酸延长基因和24.1%的三酰甘油合成基因;spl9b spl9c spl9d突变体的种子含油量显著低于野生型(Student’s t-test,P=5.4e-10,n=10),Glyma.01G075800和Glyma.11G163828敲除突变体的含油量也显著降低(Student’s t-test,P<0.01,n=5),证明SBP家族TF是调控大豆种子含油量的关键因子。
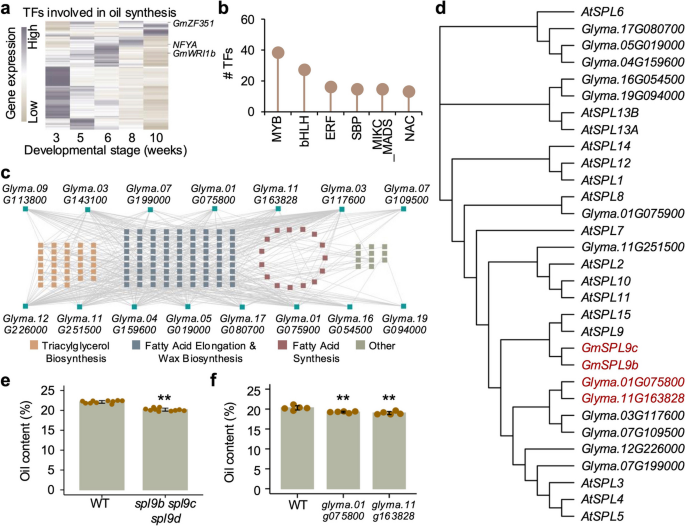
实验所用关键产品:CRISPR/Cas9载体、自主构建的大豆突变体库、95%异丙醇(油脂提取)。
3.7 利用SoyGRN定位QTL中的候选TF
实验目的是利用SoyGRN优先定位QTL中的候选TF,以干旱敏感性指数QTL为例进行验证。研究对每个QTL内的TF,计算其与其他QTL内靶基因的互作得分总和,与1000个随机选择的TF对比,筛选得分前5%的TF;对候选TF进行GO功能富集分析,验证其与性状的相关性;同时开发了在线平台SoyTFBase,供研究者查询TF-靶标互作关系。结果显示,在干旱敏感性指数QTL中,GmMYB306被鉴定为高置信度候选TF,其靶基因显著富集“响应非生物刺激”过程,且包含多个已知的干旱响应基因(如GmRVE8a),证明SoyGRN可有效突破连锁不平衡的限制,精准定位QTL中的关键调控TF;在线平台SoyTFBase支持TF靶基因查询、基因调控TF查询及多TF/基因的互作对比功能。
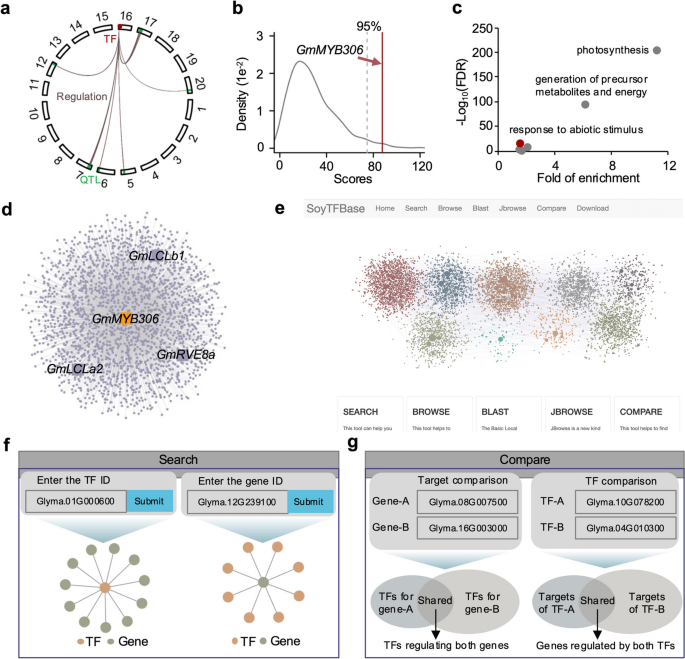
文献未提及具体实验产品,领域常规使用SoyBase等QTL注释工具及网页开发工具。
4. Biomarker研究及发现成果
本研究中鉴定的调控农艺性状的关键TF属于功能型Biomarker,包括调控种子种皮颜色的GmTT8b、调控种子含油量的SBP家族TF、调控干旱耐受性的GmMYB306。这些Biomarker的筛选逻辑为:基于SoyGRN的TF-靶标互作关系,通过超几何检验筛选与性状相关通路基因显著互作的TF,然后通过功能验证(过表达、敲除实验)确认其对性状的调控作用。
对于种皮颜色Biomarker GmTT8b,其来源为大豆基因组,验证方法包括过表达表型分析、组织表达模式分析、染色质可及性分析,结果显示过表达株系种皮颜色显著加深,且GmTT8b在种子中优先表达,其靶基因启动子在种子中染色质可及性更高;对于含油量Biomarker SBP家族TF,来源为大豆基因组,验证方法为敲除实验,结果显示spl9b spl9c spl9d突变体含油量显著降低(n=10,P=5.4e-10),Glyma.01G075800和Glyma.11G163828敲除突变体含油量也显著降低(n=5,P<0.01);对于干旱耐受性Biomarker GmMYB306,来源为大豆QTL区域,验证方法为GO功能富集分析,其靶基因显著富集干旱响应通路,且拟南芥同源基因MYB94过表达可增强干旱耐受性。
核心成果提炼:GmTT8b是首次在大豆中鉴定的调控种皮颜色的bHLH家族TF,为大豆种皮颜色的分子育种提供了新靶点;SBP家族TF是首次被报道参与大豆油脂积累的调控因子,拓展了大豆含油量调控的分子机制;GmMYB306是干旱耐受性的候选调控TF,为大豆耐旱育种提供了新的候选基因;构建的SoyGRN是当前最全面的大豆转录调控网络,为大豆功能基因组学研究提供了重要资源;开发的在线平台SoyTFBase可加速大豆农艺性状关键调控因子的挖掘,推动大豆分子育种进程。