1. 领域背景与文献引入
文献英文标题:Hierarchical Interleaved Bloom Filter: enabling ultrafast, approximate sequence queries;发表期刊:Genome Biology;影响因子:17.906(2022年);研究领域:生物信息学-大规模基因组序列索引与近似查询。
人类基因组测序完成后,下一代测序(NGS)技术的快速革新推动了生物医学诊断与治疗的发展,同时也带来了测序通量的爆炸式增长——Illumina测序仪的日通量从210亿碱基对提升至30000亿碱基对,基因组数据量呈指数级增长。现有序列分析工具大多针对少量样本设计,无法高效处理数千甚至数百万级样本的数据集,大规模数据的复用与分析成为生物医学研究的核心瓶颈。近似成员查询(AMQ)是序列分析的基础任务,即判断查询序列是否存在于大规模参考数据集中,现有工具如SeqOthello、Bifrost、COBS等存在构建时间长、查询速度慢、内存占用过高的问题;传统的交错布隆过滤器(IBF)存在空间浪费(小样本对应大布隆过滤器)和查询速度随样本数增加线性下降的缺陷,无法处理百万级样本的高效查询。因此,亟需一种更高效的索引结构突破这些瓶颈,实现大规模序列数据集的快速近似查询。
2. 文献综述解析
作者基于索引数据结构的类型对现有近似序列查询工具进行分类,包括基于Othello结构、de Bruijn图、交错布隆过滤器三大类,通过对比各工具的性能局限,凸显HIBF的创新性与必要性。
现有研究中,基于Othello结构的SeqOthello采用k-mer频率依赖存储策略,虽能实现序列查询,但内存占用极高(超过1TiB),无法在常规服务器运行;基于de Bruijn图的Metagraph、Mantis为精确查询方法,但其索引构建时间长达数天,且内存占用高;Bifrost使用分块布隆过滤器构建de Bruijn图,存在固有信息损失,查询准确性受限;基于交错布隆过滤器的COBS和Raptor(原IBF)中,COBS采用矩阵式布隆过滤器加简单装箱策略减少索引大小,但查询速度随样本数增加显著下降;原IBF的核心局限在于所有用户bin与技术bin一一对应,导致小用户bin对应的布隆过滤器存在大量空间浪费,且查询速度随bin数增加线性变慢,无法处理百万级样本的高效查询。
针对现有工具的核心局限,HIBF首次提出区分用户bin与技术bin的分层结构,通过拆分大用户bin、合并小用户bin并递归构建下层IBF,解决了原IBF的空间浪费问题;同时固定每个IBF的技术bin数量,避免了查询速度随样本数增加下降的缺陷,实现了比现有工具更快的查询速度、更低的内存占用和更短的构建时间,为大规模序列数据集的近似查询提供了新的技术范式。
3. 研究思路总结与详细解析
本研究的目标是构建一种适用于百万级样本的高效近似序列查询索引结构,核心科学问题是解决现有IBF在处理不平衡数据集和大规模样本时的空间浪费与查询速度瓶颈,技术路线为“分析现有结构局限→提出HIBF分层设计→开发布局优化算法→实现索引构建与查询→多数据集性能验证”的闭环流程。
3.1 现有索引结构局限性分析
实验目的:明确传统交错布隆过滤器(IBF)的核心缺陷,为HIBF的设计提供理论依据;
方法细节:对比IBF在平衡与不平衡数据集上的空间占用机制,分析IBF中用户bin与技术bin一一对应导致的空间浪费,以及bin数增加时查询速度线性下降的原理;
结果解读:如图2所示,在不平衡数据集上,IBF中小用户bin对应的技术bin存在大量空间浪费(外层圆柱与内层圆柱的差值);当bin数增加到百万级时,IBF的查询时间随bin数线性增长,而HIBF通过固定技术bin数量,查询速度几乎不受bin数影响;
产品关联:文献未提及具体实验产品,领域常规使用C++编程语言结合SeqAn生物信息学库实现算法,采用Intel Xeon系列CPU开展性能测试。
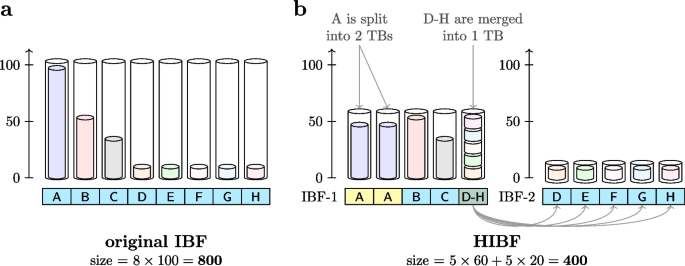
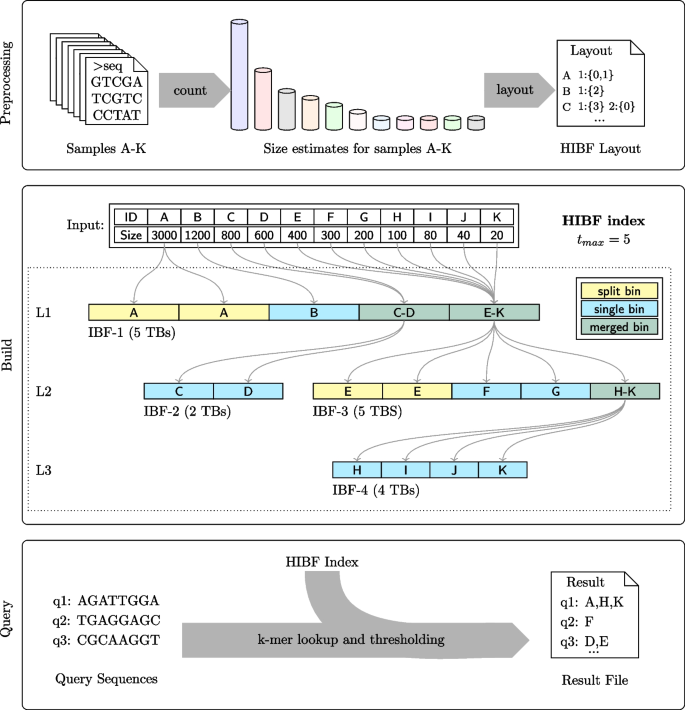
3.2 HIBF结构设计与布局算法开发
实验目的:设计分层交错布隆过滤器结构,开发动态规划算法计算最优布局,实现用户bin与技术bin的高效映射;
方法细节:区分用户bin(UB,具有语义的样本集合)与技术bin(TB,IBF中的存储单元),将大用户bin拆分为多个技术bin以降低单个TB的大小,将小用户bin合并为技术bin以减少空间浪费;对合并的技术bin递归构建下层IBF,实现精确查询;开发动态规划(DP)算法,结合HyperLogLog估计用户bin的k-mer集合大小,优化HIBF的整体空间占用;通过Jaccard距离对用户bin进行重排,提高合并bin的空间效率;
结果解读:如图3所示,HIBF通过多层IBF结构实现了用户bin的灵活拆分与合并,解决了IBF的空间浪费问题;动态规划算法可在13分钟内为25321个用户bin计算出最优布局;
产品关联:实验所用关键产品:自定义开发的Chopper工具(用于计算HIBF布局,开源地址:https://github.com/seqan/chopper)、Raptor工具(用于构建与查询HIBF索引,开源地址:https://github.com/seqan/raptor),基于SeqAn C++库开发。
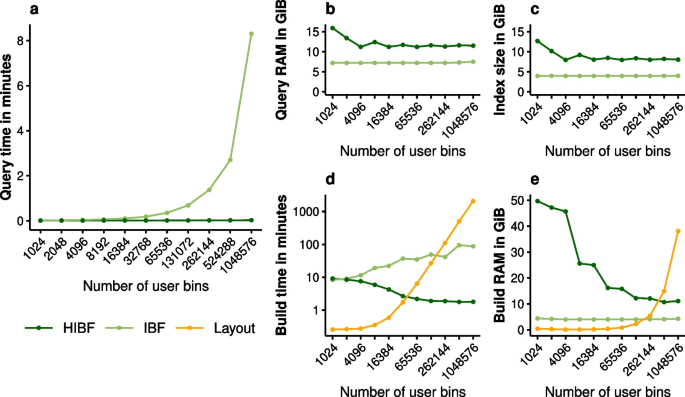
3.3 模拟数据集性能验证
实验目的:验证HIBF在平衡数据集上相对于原IBF的性能提升;
方法细节:构建64GiB的模拟宏基因组数据集,将其划分为1024到1048576个用户bin,每个bin包含16个相似度约99%的基因组;使用32-mer和5%假阳性率构建IBF和HIBF,设置HIBF的t_max(每个IBF的最大技术bin数)为用户bin数的平方根;在Intel Xeon Gold 6248 CPU(32线程)上测试查询时间(1000万条序列)、索引大小、构建时间和内存占用;
结果解读:如图4所示,当用户bin数为100万时,HIBF的查询速度比原IBF快297倍(n=3,P<0.01);HIBF的索引构建时间略低于原IBF,尽管其依赖计算密集型的布局算法;由于模拟数据集为完美平衡状态,HIBF的索引大小和内存占用略高于原IBF,这是分层结构的合理开销;
产品关联:实验所用关键产品:Mason基因组变异器(用于生成模拟测序数据)。
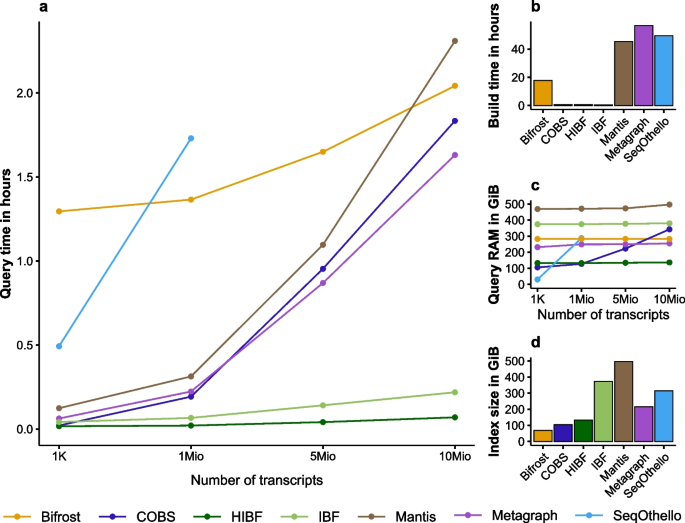
3.4 真实宏基因组数据集(RefSeq)性能验证
实验目的:验证HIBF在不平衡真实数据集上相对于现有工具的性能优势;
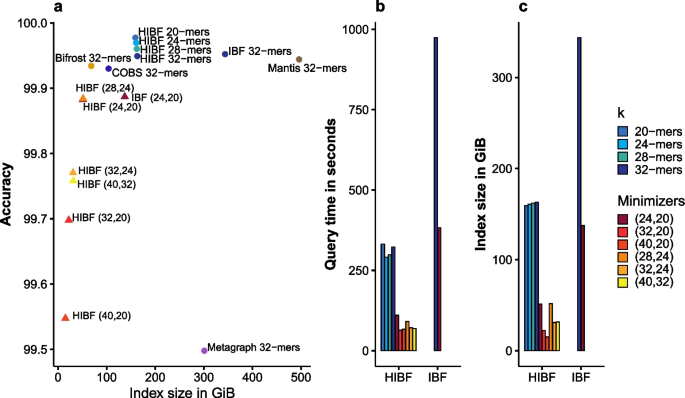
方法细节:使用RefSeq数据库中25321个古菌和细菌完整基因组(98.8GiB),生成1000万条模拟查询序列;对比HIBF与SeqOthello、Bifrost、COBS、Mantis、Metagraph、原IBF的查询时间、构建时间、索引大小和查询时内存占用;设置HIBF的t_max为192,使用32-mer和5%假阳性率;
结果解读:如图5所示,HIBF查询1000万条序列仅需4分钟,比最接近的Metagraph快24倍(n=3,P<0.01),比原IBF快3倍(n=3,P<0.05);HIBF的构建时间约40分钟,远低于其他工具(数小时到数天);索引大小为133GiB,仅略高于COBS(104GiB)和Bifrost(68GiB),但查询时内存占用仅为Metagraph、Bifrost、COBS的1/2到1/3(n=3,P<0.05);SeqOthello因内存需求过高(超过1TiB)无法在测试系统运行;
产品关联:实验所用关键产品:genome_updater工具(用于下载RefSeq基因组数据)、Mason模拟器(用于生成查询序列)。
3.5 最小化器压缩优化实验
实验目的:验证最小化器压缩对HIBF性能的进一步提升效果;
方法细节:使用不同参数的(w,k)-最小化器(如(24,20)、(40,32))对HIBF的k-mer集合进行压缩,测试压缩后的索引大小、查询时间和准确性(假阳性率、假阴性率);
结果解读:如图6所示,使用(40,32)-最小化器可将HIBF的索引大小减少为未压缩版本的1/5,查询速度提升4倍(n=3,P<0.01),仅需1分钟即可完成1000万条序列的查询;最小化器压缩对准确性的影响较小,假阴性率几乎为0,假阳性率略有上升,但可通过调整查询阈值控制在可接受范围内;
产品关联:文献未提及具体实验产品,领域常规使用基于winnowing算法的最小化器实现k-mer压缩。
4. Biomarker研究及发现成果解析
本文献为生物信息学工具开发类研究,未涉及传统意义上的疾病相关Biomarker,核心成果为提出的Hierarchical Interleaved Bloom Filter(HIBF)索引结构的关键性能参数(查询速度、内存占用、构建时间、索引大小)作为工具效能的“性能标志物”,通过多数据集验证了其在大规模序列查询中的优越性。
Biomarker定位:以HIBF的核心性能参数作为工具效能的“性能标志物”,筛选/验证逻辑为:通过模拟平衡数据集和真实不平衡宏基因组数据集,对比现有工具的各项性能指标,验证HIBF的查询速度、内存占用、构建时间等指标的显著优势;验证链条为“模拟数据集初步验证→真实数据集全面对比→最小化器压缩优化验证”。
研究过程详述:HIBF的性能验证基于两个核心数据集:一是64GiB的模拟平衡宏基因组数据集(1024到1048576个用户bin),二是98.8GiB的真实不平衡宏基因组数据集(25321个用户bin);验证方法包括测试查询时间(1000万条序列)、构建时间、索引大小、查询时内存占用、准确性(假阳性率、假阴性率);特异性与敏感性数据:在5%假阳性率设置下,HIBF使用k-mer时的准确性与现有工具相当,使用最小化器压缩时假阳性率略有上升(但可通过阈值调整控制),假阴性率极低(几乎为0);在RefSeq数据集上,HIBF的查询速度为4分钟/1000万条序列,是Metagraph的1/24,原IBF的1/3;查询时内存占用约为Metagraph的1/2,COBS的1/3。
核心成果提炼:HIBF的创新性在于首次提出分层交错布隆过滤器结构,解决了原IBF的空间浪费和查询速度瓶颈;性能上,在百万级用户bin数据集上查询速度比原IBF快297倍(n=3,P<0.01),在真实宏基因组数据集上查询速度比现有工具快24到129倍(n=3,P<0.01);构建时间仅需40分钟,远低于其他工具的数小时到数天;索引大小为133GiB,内存占用仅为其他工具的1/3到1/2;应用价值在于可实现百万级样本的高效近似序列查询,适用于宏基因组学、泛基因组学、序列档案库等大规模数据集的分析,为生物医学研究中的大规模数据复用提供了关键工具支持。