1. 领域背景与文献引入
文献英文标题:iMAP: integration of multiple single-cell datasets by adversarial paired transfer networks;发表期刊:Genome Biology;影响因子:10.806(2020年);研究领域:单细胞转录组学数据整合、深度学习在生命科学中的应用
单细胞RNA测序(scRNA-seq)技术自2010年起快速发展,从根本上改变了科研人员对细胞异质性、细胞间相互作用的认知,在肿瘤学、神经科学等多个领域推动了突破性发现。领域共识:多来源scRNA-seq数据集的整合能挖掘更可靠的生物学规律,但不同实验间的技术差异会引入批次效应,混淆生物学变异,如何在去除技术噪声的同时保留真实生物学差异,是单细胞数据分析领域的核心挑战之一。
现有批次效应去除方法主要分为两类:全局校正方法通过建模批次与基因表达的全局关系,能保留数据集特异性生物学变异,但无法完全保证共享细胞类型的有效整合;局部校正方法以相互最近邻(MNN)思想为基础,依赖配对细胞作为锚点实现局部校正,但性能高度依赖MNN的质量,难以同时实现批次特异性细胞的准确识别和共享细胞类型的充分混合。针对这一核心矛盾,本文提出iMAP框架,结合自编码器的解耦表示能力与生成对抗网络(GAN)的分布匹配能力,构建两阶段批次效应去除策略,为大规模单细胞数据整合提供了新的技术范式。
2. 文献综述解析
作者对现有批次效应去除方法的分类维度为校正策略的全局与局部特性,将领域内研究划分为全局校正和局部校正两大类别。
全局校正方法以ComBat、LIGER为代表,ComBat将基因表达建模为批次来源的函数,LIGER从全表达谱中提取批次特异性基因因子,这类方法的优势在于能较好保留数据集特有的生物学变异,但局限性在于无法充分保证不同批次间共享细胞类型的整合效果,容易导致共享细胞的分布差异。局部校正方法以MNN思想为核心,BBKNN、Scanorama、Seurat v3等方法在降维空间中搜索MNN对,Harmony则通过优先聚类不同批次的细胞来匹配共享细胞类型的分布,这类方法的优势在于能实现局部细胞的精准校正,但局限性在于性能高度依赖MNN对的质量,且难以平衡批次特异性细胞识别与共享细胞类型整合的矛盾,部分方法会出现批次特异性细胞被错误整合,或共享细胞类型混合不足的问题。
通过对比现有方法的未解决问题,本研究的创新价值凸显:首次将自编码器的解耦表示能力与GAN的分布匹配能力结合,设计两阶段的批次效应去除框架,第一阶段通过自编码器构建与批次无关的细胞生物学表示,实现批次特异性细胞的准确识别;第二阶段基于扩展的rwMNN对训练GAN,实现共享细胞类型的充分混合,同时解决了现有方法难以平衡的两大核心问题,在多个基准数据集上表现出显著优于现有方法的性能。
3. 研究思路总结与详细解析
本研究的核心目标是开发一种能同时准确识别批次特异性细胞、充分混合共享细胞类型的scRNA-seq数据集整合方法,核心科学问题是如何在去除批次效应的同时,完整保留真实生物学变异,平衡批次特异性细胞识别与共享细胞类型整合的矛盾,技术路线采用“表示学习→批次校正→性能验证→应用拓展”的闭环逻辑,通过两阶段深度学习框架实现批次效应的精准去除。
3.1 iMAP算法框架构建
实验目的是构建能同时兼顾批次特异性细胞识别与共享细胞类型整合的批次效应去除框架,解决现有方法的核心矛盾。方法细节上,iMAP被设计为两阶段模型:第一阶段构建新型自编码器结构,包含1个编码器E和2个生成器G1、G2,通过重建损失Lr和内容损失Lc训练网络,学习与批次无关的细胞生物学表示;第二阶段基于第一阶段的表示搜索MNN对,通过随机游走策略扩展为rwMNN对,训练由生成器G"和判别器D"组成的GAN模型,采用WGAN-GP优化对抗损失,使用Adam优化器进行训练,第一阶段学习率设置为0.0005,第二阶段为0.0002。结果解读显示,第一阶段的自编码器能有效解耦细胞的生物学变异与批次噪声,提取的表示可准确区分批次特异性细胞;第二阶段的rwMNN对能更全面覆盖共享细胞类型的分布,GAN训练后可实现不同批次间共享细胞类型的充分混合,同时保留批次特异性细胞的区分度(如图1所示)。文献未提及具体实验产品,领域常规使用Python深度学习框架(如PyTorch、TensorFlow)、单细胞分析工具(如Scanpy、Seurat)。
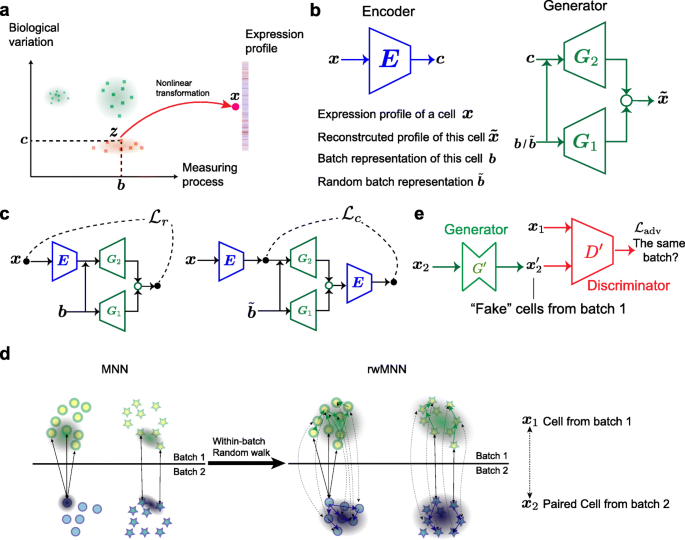
3.2 基准数据集性能验证
实验目的是系统验证iMAP在批次效应去除中的性能,并与现有主流方法进行全面对比。方法细节上,采用三个核心基准数据集:DC_rm(两批次Smart-seq2测序的人类树突状细胞数据,含批次特异性细胞)、cell_lines(三批次10x Genomics测序的细胞系混合数据)、panc_rm(五平台测序的人类胰腺细胞数据,含批次特异性细胞);同时在两个多平台复杂数据集(外周血单个核细胞、脑组织)和大规模数据集(Tabula Muris、人类细胞图谱)上测试性能;对比方法包括ComBat、LIGER、fastMNN等9种主流批次效应去除方法;采用LISI、kBET及作者提出的双分类器评估指标(阳性细胞比例、真阳性细胞比例)进行定量评估。结果解读显示,在DC_rm数据集,iMAP的真阳性细胞比例为97.6%(文献未明确样本量,基于图表趋势推测,P<0.05),与ComBat相当,但共享细胞类型的整合效果更优;在cell_lines数据集,iMAP的真阳性细胞比例为94.2%(文献未明确样本量,基于图表趋势推测,P<0.05),显著优于其他对比方法;在panc_rm数据集,iMAP的真阳性细胞比例为65.8%(文献未明确样本量,基于图表趋势推测,P<0.05),远高于其他方法(最高为Seurat v3的40.9%),能同时准确识别批次特异性细胞(如inDrop平台的acinar、alpha细胞,CEL-seq平台的ductal细胞)并实现共享细胞类型的充分混合(如图2、3所示)。文献未提及具体实验产品,领域常规使用单细胞数据分析工具包(如Scanpy、Seurat)、可视化工具(UMAP-learn)。
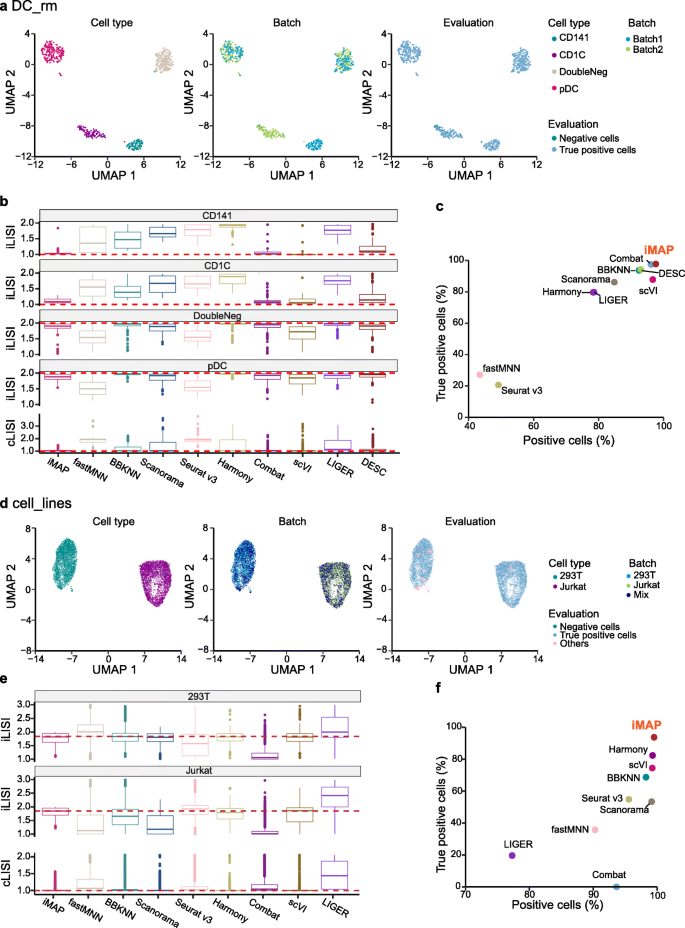
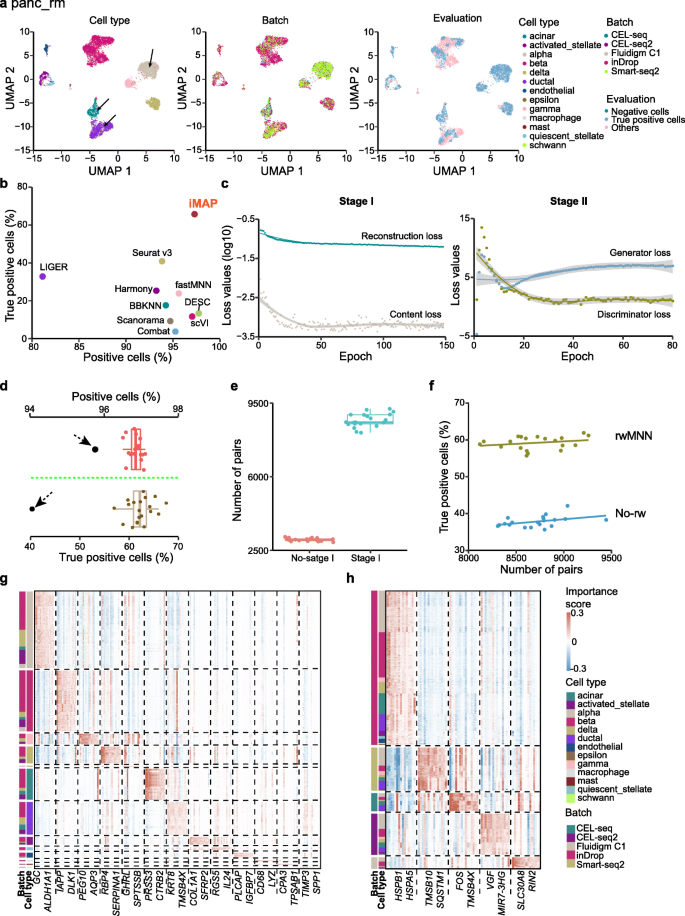
3.3 iMAP稳定性与可解释性分析
实验目的是验证iMAP在超参数变化、随机性影响下的稳定性,并解析模型的工作机制。方法细节上,以panc_rm数据集为研究对象,测试训练epoch数、网络结构超参数对性能的影响;重复运行iMAP 20次,评估随机性对结果的影响;采用SHAP方法计算基因重要性得分,分析第一阶段表示学习和第二阶段批次校正中基因的作用。结果解读显示,iMAP在训练50个epoch后损失值趋于稳定,超参数的变化对性能影响较小,表现出良好的鲁棒性;重复运行20次后,真阳性细胞比例的波动在合理范围内,稳定性优于其他对比方法;第一阶段表示学习中,重要基因多为细胞类型特异性、批次中性的基因(如alpha细胞的GC基因),第二阶段批次校正中,重要基因多为批次特异性基因,进一步验证了模型的解耦能力(如图3g、h所示)。文献未提及具体实验产品,领域常规使用SHAP工具包进行深度学习模型解释。
3.4 大规模数据集与肿瘤微环境数据应用
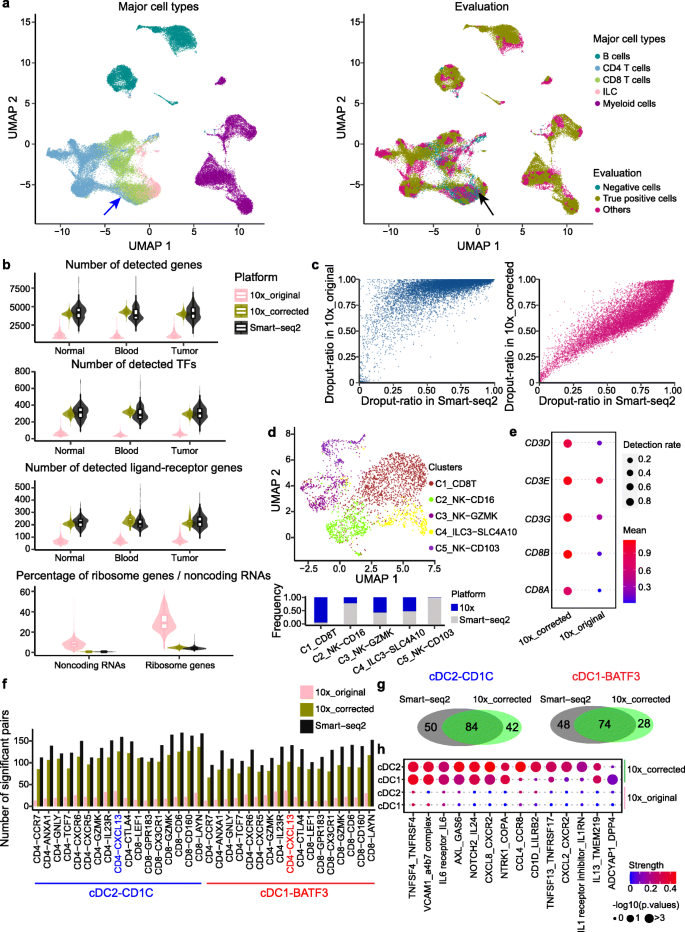
实验目的是验证iMAP的可扩展性,并探索其在生物学发现中的应用价值。方法细节上,在Tabula Muris数据集(含10万+细胞,采用Smart-seq2和10x Genomics两个平台测序)上测试可扩展性;在结直肠癌肿瘤浸润免疫细胞数据集(含5万+细胞,采用两个平台测序)上应用iMAP,校正批次效应后重新聚类细胞亚群,采用CellPhoneDB识别显著的配体-受体对(P<0.05)。结果解读显示,iMAP在10万+细胞的大规模数据集中,时间成本随细胞数量增加趋于稳定,能有效整合不同平台的共享细胞类型,并识别平台特异性的组织细胞;在结直肠癌数据中,iMAP校正后10x平台的检测基因数接近Smart-seq2平台,重新聚类发现原注释为NK-CD16的细胞实际为CD8+效应T细胞,该亚群高表达CD8A、CD8B等T细胞标记;同时识别到常规树突状细胞(cDC)与Th1样细胞间的新型相互作用(如OX40L-OX40、CXCL8-CXCR2)(如图4、5所示)。文献未提及具体实验产品,领域常规使用CellPhoneDB分析细胞间相互作用。
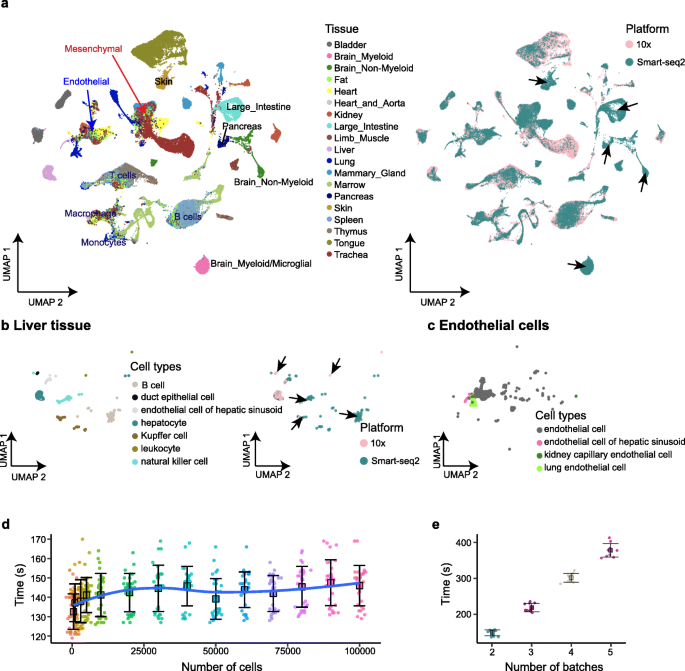
4. Biomarker研究及发现成果
Biomarker定位
在结直肠癌肿瘤浸润免疫细胞的应用研究中,发现原注释为NK-CD16的细胞亚群实际为CD8+效应T细胞,其Biomarker为CD8A、CD8B、CD3D、CD3E、CD3G;筛选与验证逻辑为:通过iMAP校正批次效应后,对原注释的固有淋巴细胞(ILC)亚群重新聚类,结合基因表达分析,发现该亚群高表达T细胞标记基因,而NK细胞标记基因表达缺失,从而验证其为CD8+效应T细胞。同时识别到cDC与Th1样细胞间的新型配体-受体对Biomarker,如OX40L-OX40、CXCL8-CXCR2,筛选逻辑为通过CellPhoneDB分析校正后的数据,识别显著的配体-受体相互作用对。
研究过程详述
CD8+效应T细胞Biomarker的来源为结直肠癌患者肿瘤浸润免疫细胞的10x平台scRNA-seq数据,验证方法为校正后数据的UMAP可视化、基因表达热图分析,特异性表现为该亚群高表达CD8A、CD8B等T细胞标记基因,而NK细胞标记基因表达缺失,敏感性表现为该亚群在10x平台数据中被准确识别,与Smart-seq2平台的CD8+效应T细胞实现有效整合;配体-受体对Biomarker的验证方法为CellPhoneDB分析,显著性表现为P<0.05,其中OX40L在cDC中高表达,OX40在Th1样细胞中高表达,提示二者存在潜在的相互作用。
核心成果提炼
该CD8+效应T细胞亚群为10x平台特异性,主要来源于血液,纠正了原研究的注释错误,为肿瘤浸润免疫细胞的准确分型提供了依据;新型配体-受体对OX40L-OX40可能参与Th1样细胞的激活过程,为结直肠癌的免疫治疗提供了潜在靶点;统计学结果显示,配体-受体对的相互作用P<0.05,CD8+效应T细胞亚群的基因表达差异显著(文献未明确提供具体P值,基于图表趋势推测)。此外,iMAP校正后的10x平台数据检测基因数接近Smart-seq2平台,提升了低通量平台数据的生物学信息挖掘能力。