1. 领域背景与文献引入
文献英文标题:Quantitative principles of cis-translational control by general mRNA sequence features in eukaryotes;发表期刊:Genome Biology;影响因子:未公开;研究领域:真核生物mRNA顺式翻译调控
真核生物基因表达调控是生命科学领域的核心研究方向,翻译作为基因表达的关键环节,其调控机制直接决定蛋白质的合成效率和细胞功能。翻译调控主要分为顺式作用元件和反式作用因子介导的调控,其中顺式作用元件又可分为通用序列特征和基因/条件特异性元件。通用序列特征包括mRNA 5"区域二级结构、上游开放阅读框(uORF)、起始AUG侧翼序列、编码区(CDS)长度及密码子使用模式,这些特征广泛存在于所有或多数基因中,在多种生理状态下发挥作用;而特异性元件仅存在于部分基因中,由miRNA或特异性RNA结合蛋白等反式因子调控。
领域共识:此前的研究已证实通用序列特征会影响单个基因的翻译速率,但它们对全基因组翻译速率变异的定量贡献尚未得到系统解析,不同真核生物物种间的调控共性与差异也缺乏深入比较,这限制了对真核生物翻译调控机制的全面理解。针对这一研究空白,本研究通过优化统计模型,系统分析五个真核模式生物(酿酒酵母、裂殖酵母、拟南芥、小鼠、人类)的核糖体图谱数据,旨在量化通用mRNA序列特征对翻译速率变异的贡献,解析其调控机制及跨物种保守性,为真核生物翻译调控研究提供更精准的定量视角。
2. 文献综述解析
本文的文献综述部分以顺式作用元件的类型为核心分类维度,将真核生物翻译调控元件分为通用序列特征和基因/条件特异性元件两类,系统梳理了现有研究的进展与局限性。
现有研究已明确通用序列特征中的每个单独元件(如mRNA二级结构、uORF)均可影响翻译速率,部分研究在酿酒酵母和哺乳动物中分析了部分特征与翻译速率的相关性,证实核糖体图谱数据可有效估计全基因组翻译速率;这些研究的技术优势在于能在全基因组水平开展高通量分析,但局限性也较为明显:多数研究依赖平均折叠能或固定窗口分析RNA二级结构,未能精准捕捉关键调控区域;对通用特征的联合贡献分析不足,且缺乏跨物种的系统比较,无法揭示调控规律的共性与差异;同时,对不同调控过程和mRNA区域之间的协同作用也未深入探讨。
通过对比现有研究的未解决问题,本研究的创新价值凸显:首次系统分析五个真核生物的通用顺式元件对翻译速率变异的联合贡献,优化RNA二级结构的预测模型,发现最折叠的25-60nt片段是关键调控区域;揭示不同物种间顺式调控的共性和差异,证实非哺乳动物物种的起始AUG近端元件(APE)调控作用更强;发现不同生化过程和mRNA不同区域之间的共线性调控,解释其减少核糖体碰撞、提高翻译机器使用效率的生物学意义,为真核生物翻译调控机制提供了全新的定量框架。
3. 研究思路总结与详细解析
本研究以量化通用mRNA序列特征对真核生物翻译速率变异的贡献为核心目标,围绕“通用顺式元件如何协同调控翻译速率、跨物种调控规律有何异同”的科学问题,采用“数据集选择→特征构建→模型优化→结果分析→结论总结”的闭环技术路线,通过五个真核模式生物的系统分析,揭示了顺式翻译调控的定量规律和共线性调控的生物学意义。
3.1 数据集选择与预处理
实验目的:获取具有代表性的真核生物翻译速率数据,排除实验干扰因素,确保分析结果的可靠性。
方法细节:选择五个模式生物的核糖体图谱数据集,其中部分物种补充不同组织或生物学重复的数据以验证结果的稳健性;对每个基因选择代表性mRNA异构体,去除mRNA丰度<1 RPKM的低丰度转录本;针对拟南芥和小鼠中存在的多mRNA异构体现象,筛选仅表达单个异构体的基因进行重复分析,排除异构体复杂性对结果的影响。
结果解读:不同物种的5"非翻译区(UTR)长度差异显著,酿酒酵母的平均5"UTR长度最短,哺乳动物和裂殖酵母的最长;而CDS长度的范围在五个物种中较为一致;所有物种的翻译速率变异范围相对较窄;单异构体基因的分析结果与所有基因的结果无显著差异,说明mRNA异构体复杂性不会影响本研究的核心结论。
产品关联:文献未提及具体实验产品,领域常规使用核糖体图谱测序技术获取翻译速率数据、ViennaRNA RNAfold软件进行RNA二级结构预测、R语言开展统计分析与模型构建。
3.2 RNA二级结构调控分析
实验目的:解析mRNA 5"区域二级结构对翻译速率的调控作用,确定关键调控区域的结构特征。
方法细节:使用ViennaRNA RNAfold软件预测mRNA 5"区域(从5"帽到起始AUG下游+35位)的Gibbs自由能;构建多种基于折叠能的特征,包括单窗口(长度6-100nt,步长1nt)、多窗口统计量(平均能、极值能、阈值占比等)及全区域折叠能;采用贝叶斯信息准则(BIC)筛选最优特征集(RNAfold模型),分析最折叠窗口的碱基配对数、茎环结构等特征与翻译速率的关联。
结果解读:RNAfold模型可解释11-33%的翻译速率变异,预测能力是之前模型的两倍以上;最折叠的25-60nt片段对翻译速率的调控作用远强于其他区域,其碱基配对数与翻译速率呈负相关,即碱基配对越多,翻译速率越低;长程碱基配对(间隔>60nt)对翻译的调控作用极小,说明局部二级结构是主要调控因素;不同物种的最优最折叠窗口长度存在差异,酵母为25-35nt,哺乳动物为55-60nt。
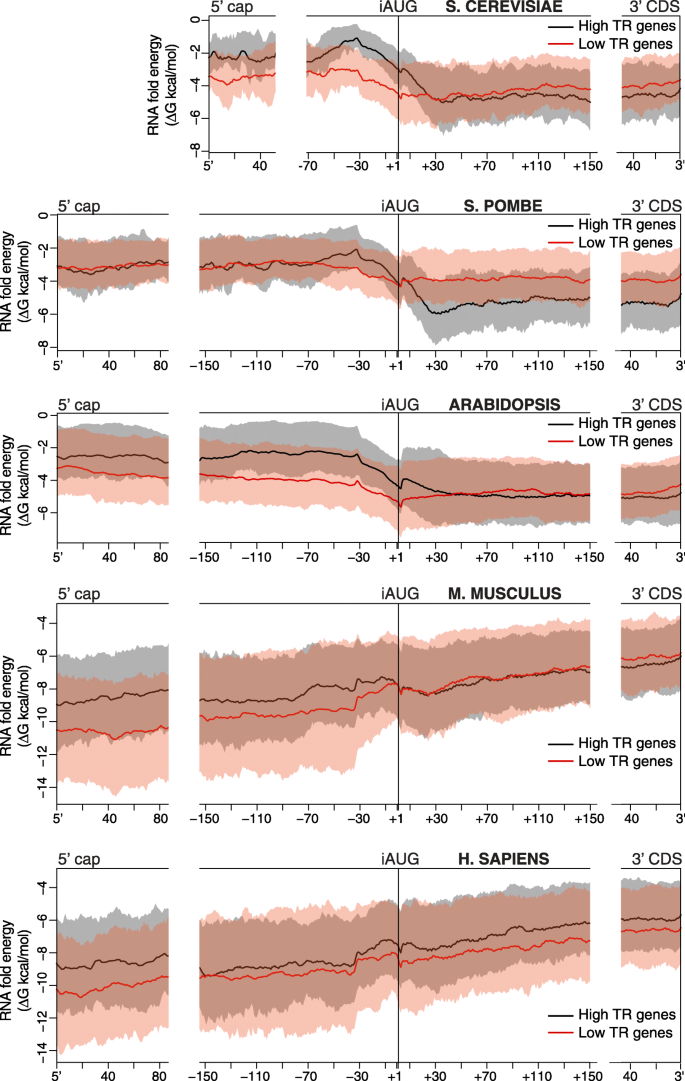
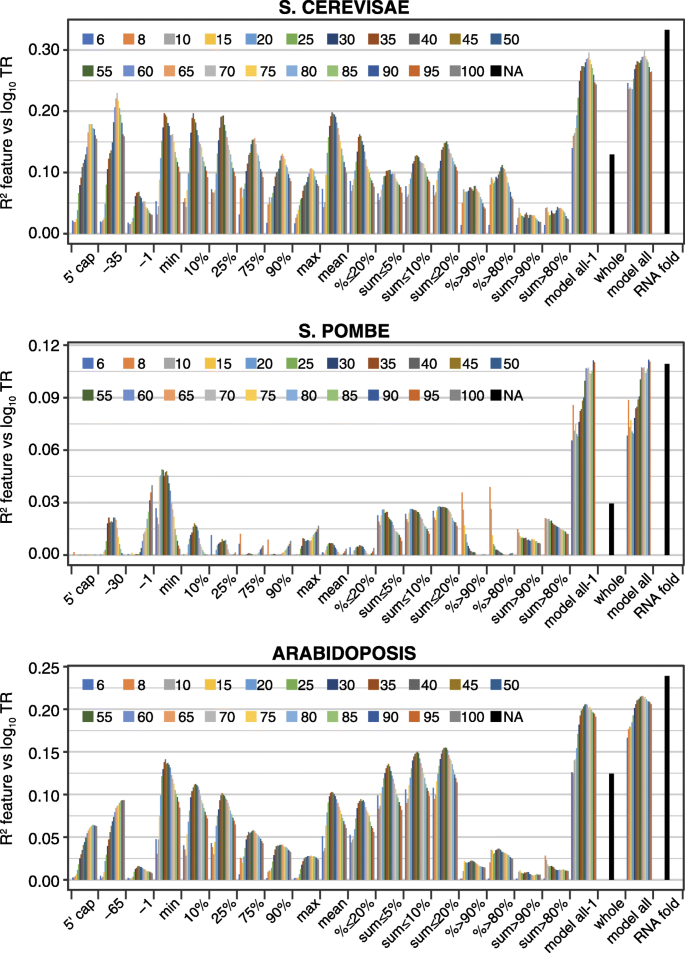
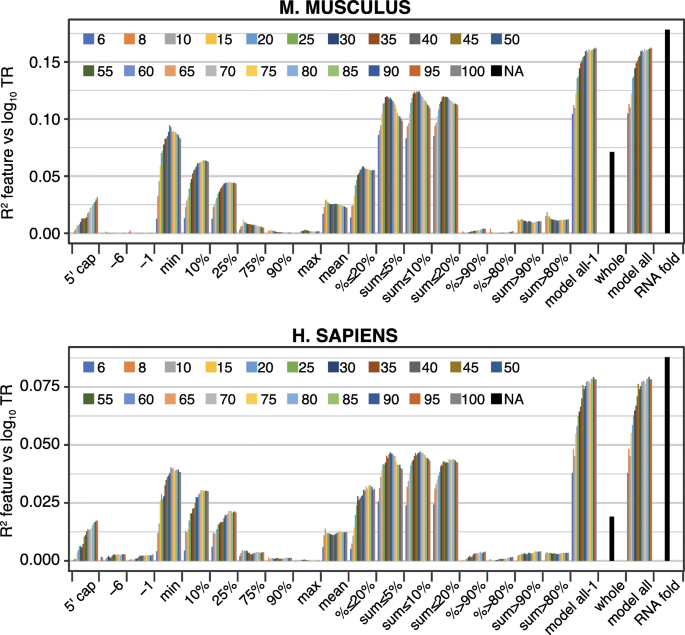
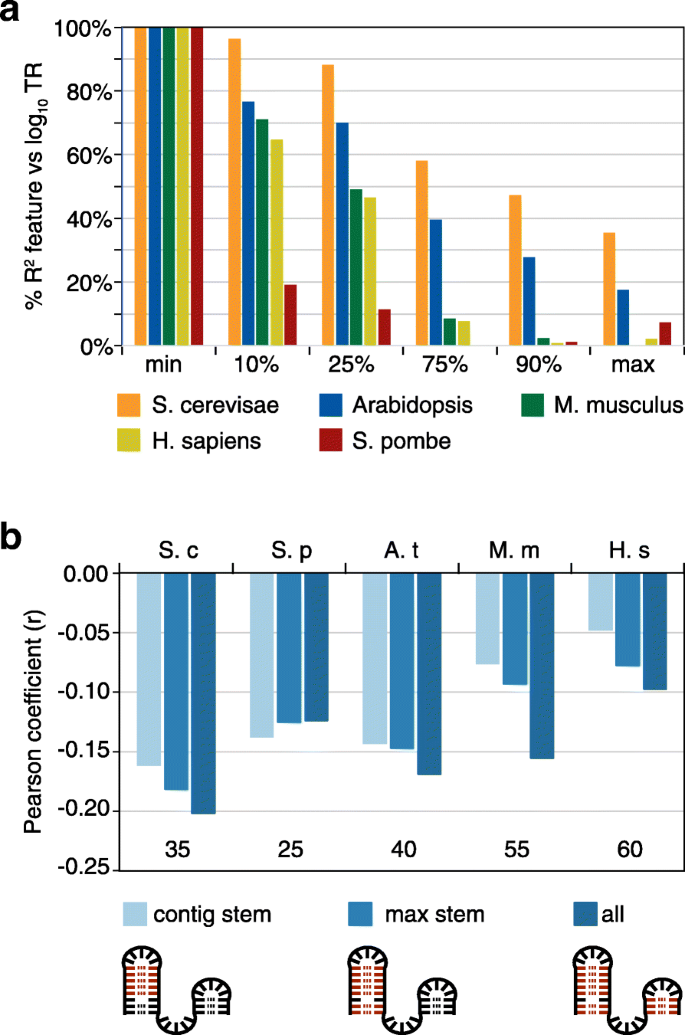
产品关联:文献未提及具体实验产品,领域常规使用ViennaRNA软件包进行RNA二级结构预测、R语言的统计分析包开展模型构建与特征选择。
3.3 5"区域序列基序调控分析
实验目的:解析mRNA 5"区域序列基序对翻译速率的调控作用,比较跨物种的基序保守性与特异性。
方法细节:以翻译速率最高的10%基因为样本,构建位置权重矩阵(PWM)确定起始AUG近端元件(APE)的最优边界;结合二核苷酸和三核苷酸频率特征,采用BIC筛选最优特征集(5"motifs模型);计算高/低翻译速率基因的三核苷酸频率比,分析mRNA不同区域(5"ofAPE、uAPE、dAPE、CDS 3"ofAPE)的基序相关性。
结果解读:非哺乳动物物种(酿酒酵母、裂殖酵母、拟南芥)的APE对翻译速率的调控作用更强,可解释16-33%的翻译速率变异,而哺乳动物仅能解释3-4%;跨物种的5"区域三核苷酸频率比存在显著正相关,说明顺式调控序列具有进化保守性;uORF的AUG基序在低翻译速率基因中显著富集,体现其对主开放阅读框翻译的抑制作用。
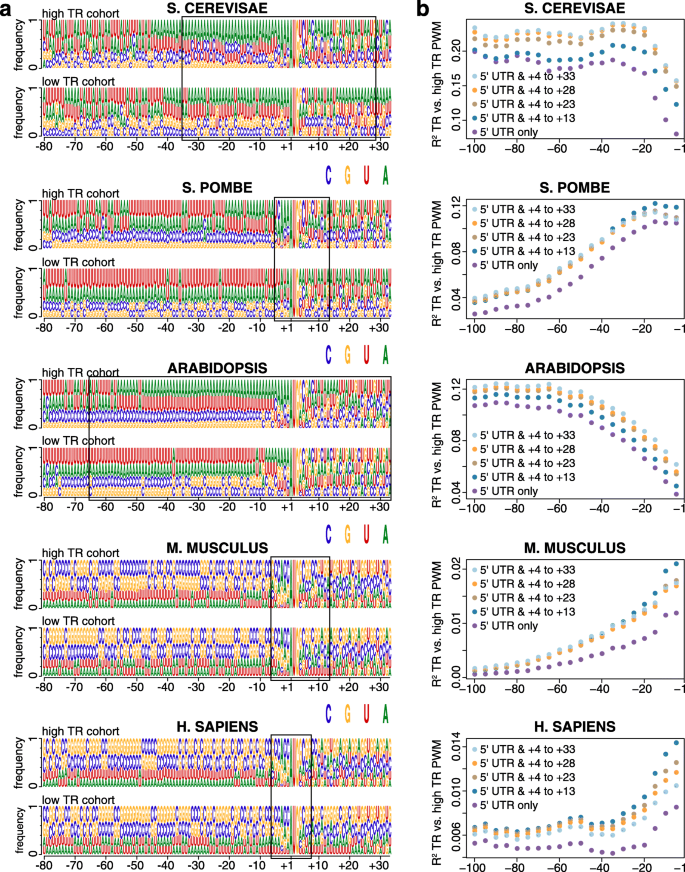
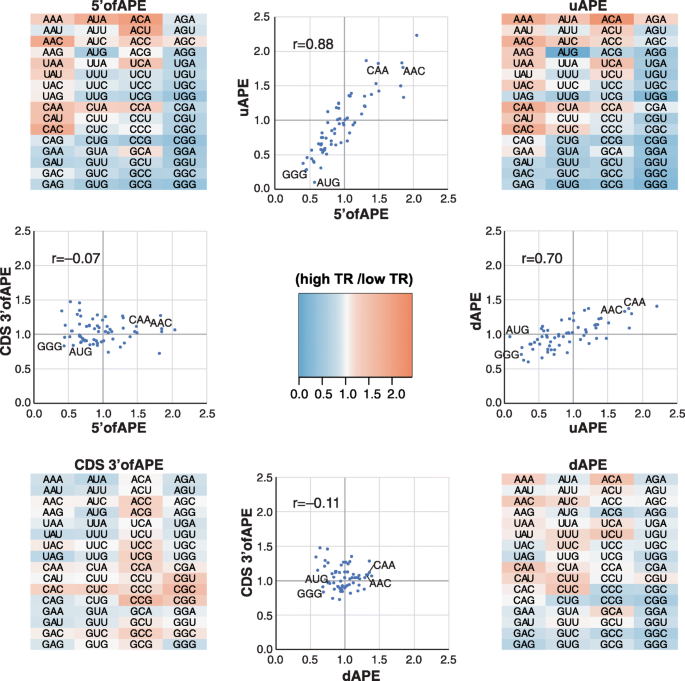
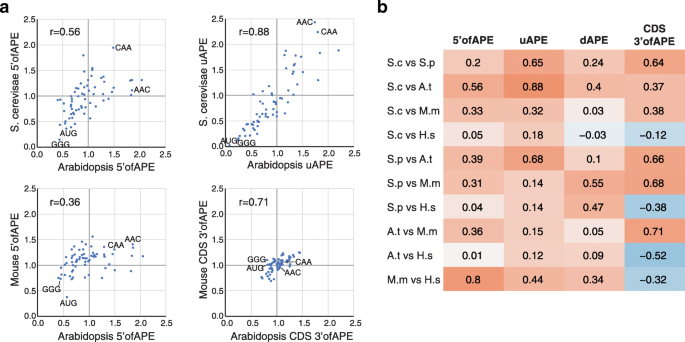
产品关联:文献未提及具体实验产品,领域常规使用序列分析软件(如MEME Suite)开展基序预测、R语言进行频率统计与模型构建。
3.4 通用特征的联合贡献与共线性调控分析
实验目的:量化所有通用特征对翻译速率变异的联合贡献,解析不同调控过程和mRNA区域之间的协同作用。
方法细节:构建包含RNAfold、5"motifs、uAUG、CDS长度、密码子使用的多元线性模型,计算联合模型的R2值以评估总贡献;通过计算特征间的R2重叠率分析不同调控过程的共线性;分析mRNA不同区域(如最折叠窗口与次折叠窗口、CDS的N端与C端)的调控相关性,验证共线性调控的存在。
结果解读:通用特征的联合模型可解释37-81%的翻译速率变异,其中酿酒酵母的解释度最高(81%),哺乳动物最低(42-46%);不同特征之间存在显著共线性,酵母中5/6对特征的重叠率达47-100%,哺乳动物为22-59%;mRNA不同区域的调控也存在强共线性,次折叠窗口与最折叠窗口的调控重叠率达63-98%,CDS的N端与C端密码子使用的调控重叠率达49-85%;共线性调控的生物学意义在于使翻译复合物沿mRNA的分布更均匀,减少核糖体碰撞,提高翻译机器的使用效率。
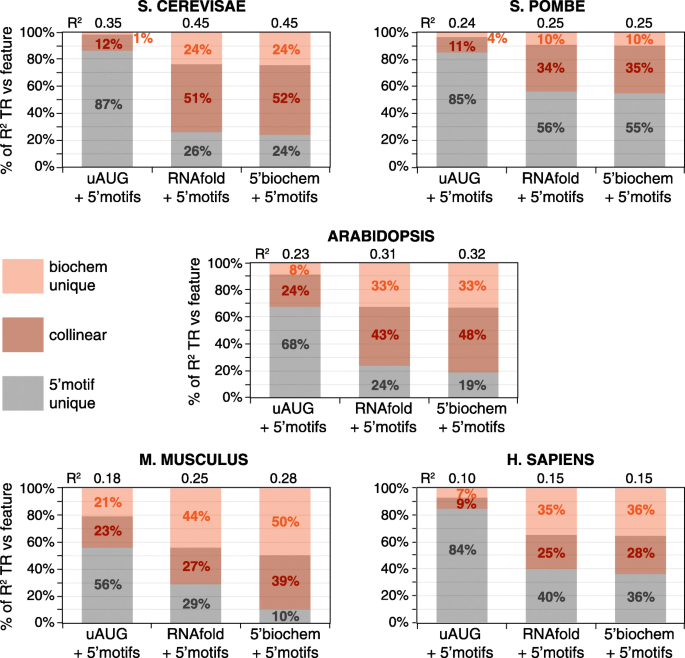
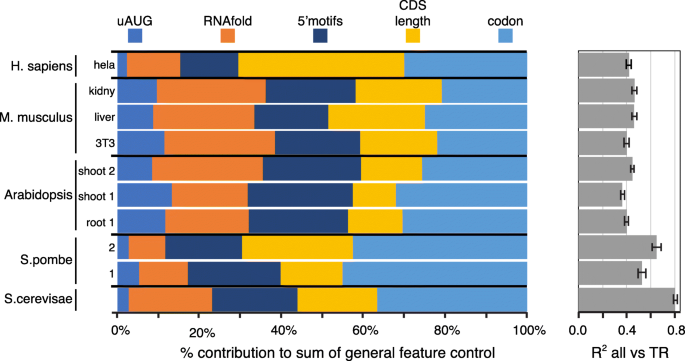
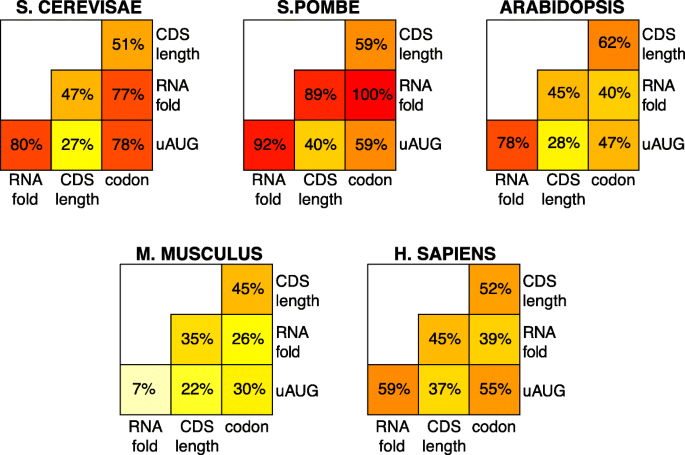
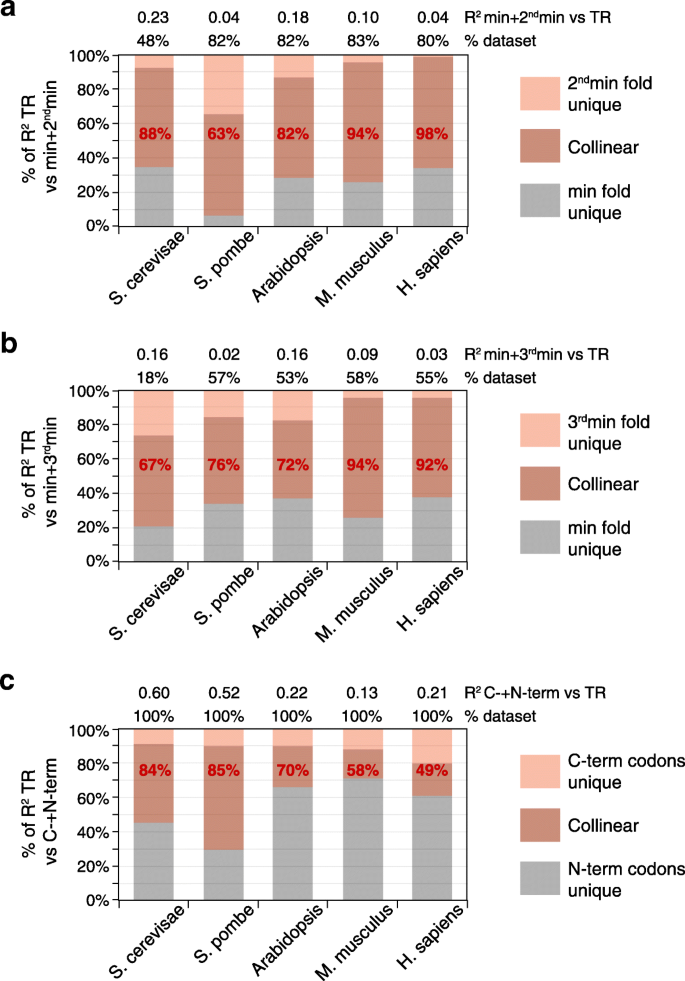
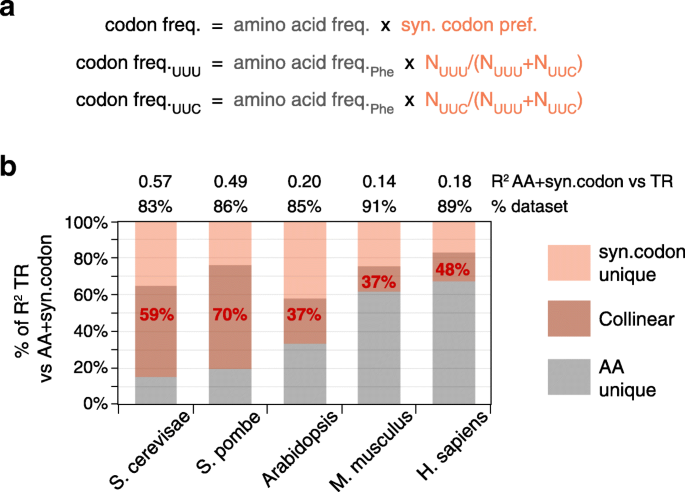
产品关联:文献未提及具体实验产品,领域常规使用R语言进行多元线性回归、共线性分析与可视化。
4. Biomarker研究及发现成果
本文将调控真核生物翻译速率的通用mRNA序列特征作为功能性生物标志物,通过全基因组关联分析和跨物种验证,明确了其调控作用、保守性和生物学意义,为真核生物翻译调控研究提供了关键的定量依据。
Biomarker定位:本文的功能性Biomarker包括真核生物中调控翻译速率的五类通用mRNA序列特征:5"区域最折叠的25-60nt片段、起始AUG近端元件(APE)、上游开放阅读框(uORF)、CDS长度、密码子使用模式。筛选/验证逻辑为:通过全基因组核糖体图谱数据关联分析筛选与翻译速率相关的序列特征→构建统计模型优化特征以提高预测能力→跨五个真核模式生物验证调控规律的共性与差异→通过共线性分析揭示其协同调控的生物学意义,形成完整的筛选-验证闭环。
研究过程详述:Biomarker来源为五个真核模式生物的全基因组mRNA序列和核糖体图谱数据;验证方法包括多元线性回归模型(以R2值评估预测能力)、BIC特征选择、跨物种序列比对、共线性分析;特异性与敏感性方面,RNAfold模型可解释11-33%的翻译速率变异,5"motifs模型在非哺乳动物中可解释16-33%的变异,联合模型可解释37-81%的变异(n=各物种基因数,模型的95%置信区间较窄,说明结果稳健)。
核心成果提炼:首次明确通用mRNA序列特征对真核生物翻译速率变异的贡献远高于此前的估计,酵母中最高达81%;发现5"区域最折叠的25-60nt片段是RNA二级结构调控的关键区域,其碱基配对数与翻译速率负相关;揭示跨物种的顺式调控序列保守性,非哺乳动物的APE调控作用更强;发现不同生化过程和mRNA区域之间的共线性调控,其生物学意义在于减少核糖体碰撞、提高翻译机器使用效率;这些成果为真核生物翻译调控机制提供了精准的定量模型,为基因表达调控的研究和应用提供了新的方向。