1. 领域背景与文献引入
文献英文标题:Scl-Ab reverts pro-osteoclastogenic signalling and resorption in estrogen deficient osteocytes;发表期刊:BMC Molecular and Cell Biology;影响因子:未公开;研究领域:骨生物学/绝经后骨质疏松症靶向治疗
绝经后骨质疏松症是女性绝经后高发的骨骼退行性疾病,核心病理机制为雌激素缺乏导致破骨细胞骨吸收活性异常增强,骨重塑失衡引发骨量快速丢失与骨折风险显著升高。传统抗吸收疗法如双膦酸盐通过直接抑制破骨细胞功能发挥作用,但临床数据显示其仅能降低约50%的骨折易感性,仍存在未被满足的治疗需求。近年来,以硬骨素(sclerostin)中和抗体(Scl-Ab,以下简称硬骨素抗体)为代表的促骨形成合成疗法成为研究热点,动物实验与临床试验均证实硬骨素抗体可显著诱导骨形成、增加骨量与骨强度,大幅降低骨折风险。
骨细胞作为骨骼中的主要机械感受器,通过分泌核因子κB受体活化因子配体(RANKL)、骨保护素(OPG)等细胞因子调控骨重塑,且硬骨素已被证实可诱导骨细胞合成核因子κB受体活化因子配体,但硬骨素抗体如何调控骨细胞对破骨细胞分化与功能的具体机制尚未完全阐明。此前研究发现,雌激素缺乏状态下成骨细胞与骨细胞的破骨生成信号(核因子κB受体活化因子配体/骨保护素比值)会发生改变,成骨细胞诱导的破骨生成与骨吸收作用显著加剧,但雌激素缺乏骨细胞是否直接参与并加剧破骨生成过程仍属研究空白。针对这一领域缺口,本研究旨在从体外层面明确两个核心科学问题:一是验证雌激素缺乏时骨细胞是否可直接诱导破骨生成与骨吸收;二是探究硬骨素抗体是否可通过调控骨细胞中核因子κB受体活化因子配体/骨保护素的表达水平,逆转骨细胞介导的破骨生成与骨吸收作用,为硬骨素抗体的临床应用机制提供细胞层面的实验依据。
2. 文献综述解析
作者围绕绝经后骨质疏松症的治疗策略、骨细胞在骨重塑中的调控作用、雌激素缺乏对骨细胞功能的影响及硬骨素抗体的作用机制四大维度,对现有研究进行系统性梳理与评述,明确了领域内的研究进展与未解决问题。
现有研究可分为三类,第一类是抗吸收与促骨形成疗法的疗效对比研究,这类研究证实抗吸收疗法虽能抑制破骨细胞活性,但对骨形成无促进作用,疗效存在局限性;而硬骨素抗体等促骨形成疗法可同时促进骨形成与抑制骨吸收,在动物与临床研究中展现出更优的骨折风险降低效果。第二类是骨细胞的骨重塑调控机制研究,这类研究明确骨细胞是骨骼中感知机械负荷的核心细胞,通过分泌核因子κB受体活化因子配体与骨保护素调控破骨细胞的分化与功能,核因子κB受体活化因子配体/骨保护素比值是决定骨吸收活性的关键指标;同时,雌激素缺乏会影响骨细胞的机械感知功能,改变其核因子κB受体活化因子配体/骨保护素的表达水平,但此类研究多聚焦于成骨细胞,对骨细胞直接介导的破骨生成作用关注不足。第三类是硬骨素抗体的作用机制研究,这类研究证实硬骨素抗体可通过抑制硬骨素对Wnt通路的拮抗作用促进骨形成,但关于硬骨素抗体如何调控骨细胞与破骨细胞间信号传导的研究仍较为匮乏。现有研究的技术方法优势在于部分研究采用了骨细胞系与机械刺激模型模拟体内环境,但局限性也较为明显,如多数研究使用的MLO-Y4骨细胞系硬骨素表达量极低,难以精准研究硬骨素相关的信号通路;且缺乏针对雌激素缺乏骨细胞与破骨细胞间直接相互作用的体外实验验证。
本研究通过对比现有研究的缺口,首次聚焦雌激素缺乏骨细胞对破骨生成的直接调控作用,采用高硬骨素表达的OCY454骨细胞系与振荡流体流机械刺激模型,精准模拟体内骨细胞的生理状态,明确了雌激素缺乏骨细胞可通过上调核因子κB受体活化因子配体/骨保护素比值与趋化因子表达增强破骨生成与骨吸收,同时证实硬骨素抗体可通过逆转上述基因表达变化抑制破骨生成,填补了领域内关于骨细胞在雌激素缺乏状态下破骨调控机制的研究空白,为硬骨素抗体的临床疗效提供了细胞层面的直接实验依据。
3. 研究思路总结与详细解析
本研究以“雌激素缺乏骨细胞对破骨生成的调控作用及硬骨素抗体的干预机制”为核心科学问题,采用“细胞模型构建→机械刺激模拟体内环境→条件培养基/共培养实验验证功能→分子水平解析信号通路”的闭环技术路线,通过体外实验系统验证了研究假设,明确了硬骨素抗体逆转雌激素缺乏骨细胞促破骨生成作用的分子机制。
3.1 骨细胞与破骨前体细胞模型构建及处理
实验目的:构建雌激素缺乏的骨细胞模型,模拟绝经后女性骨细胞的生理状态,同时施加硬骨素抗体干预,为后续功能与机制研究奠定基础。
方法细节:采用OCY454骨细胞系(可分泌高浓度硬骨素,适合硬骨素相关机制研究),先在33℃条件下扩增培养,随后转移至37℃半许可温度下分化培养15天;将分化后的骨细胞分为雌激素处理组(E组,10nM 17β-雌二醇处理6天)与雌激素缺乏组(ED组,先经10nM 17β-雌二醇处理3天,随后撤去雌激素继续培养3天);同时,为验证硬骨素抗体的作用,在两组中分别设置硬骨素抗体处理亚组(300ng/ml硬骨素抗体处理24小时)与对照组(PBS处理);所有骨细胞在实验前均采用平行板流室施加1Pa剪切应力、0.5Hz频率的振荡流体流刺激1小时,模拟体内骨细胞所受的机械负荷;破骨前体细胞采用两种模型,分别为从8月龄雌性C57BL/6小鼠骨髓中分离的原代骨髓巨噬细胞(BMM),以及RAW264.7巨噬细胞系。
结果解读:成功构建了雌激素缺乏的骨细胞模型,机械刺激处理模拟了体内骨细胞的力学微环境,为后续研究骨细胞与破骨前体细胞的相互作用提供了生理相关性的实验体系。
产品关联:实验所用关键产品:硬骨素抗体(Scl-Ab VI)由UCB Pharma/Amgen Inc.提供;RNA提取采用罗氏High pure RNA isolation kit,逆转录采用QuantiNova Reverse Transcription Kit,实时荧光定量PCR(qRT-PCR)采用QuantiNova SYBR Green PCR Kit;抗酒石酸酸性磷酸酶(TRAP)染色使用商用抗酒石酸酸性磷酸酶染色试剂盒。
3.2 条件培养基实验验证骨细胞对破骨生成与骨吸收的调控作用
实验目的:通过非接触式的条件培养基实验,验证雌激素缺乏骨细胞分泌的可溶性因子是否可增强破骨前体细胞的分化与骨吸收功能,以及硬骨素抗体是否可逆转该作用。
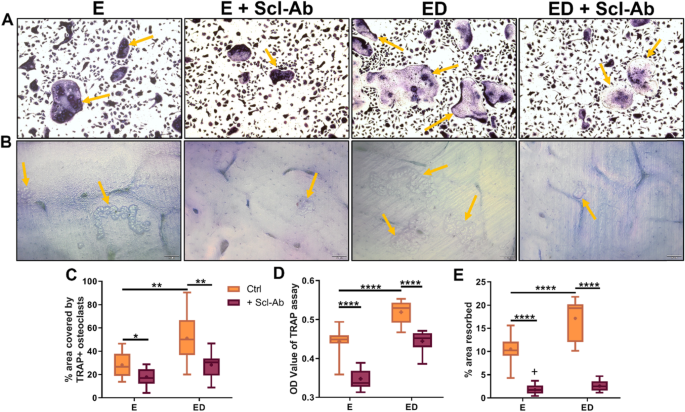
方法细节:收集不同处理组骨细胞经机械刺激24小时后的条件培养基(CM),将BMM或RAW264.7细胞接种于96孔板,加入50%对应组别的CM与50%完全培养基,并添加15ng/ml核因子κB受体活化因子配体诱导破骨分化;培养5天后进行抗酒石酸酸性磷酸酶染色,计数抗酒石酸酸性磷酸酶阳性多核细胞比例,同时检测培养上清中的抗酒石酸酸性磷酸酶活性;采用牛骨片进行骨吸收实验,将BMM接种于牛骨片上,加入50% CM与15ng/ml核因子κB受体活化因子配体培养10天(培养第7天用盐酸将培养基pH调至6.9以模拟骨吸收所需的酸性环境),随后去除细胞,经甲苯胺蓝染色后观察并定量骨吸收陷窝面积。
结果解读:与E组相比,ED组CM处理的BMM中抗酒石酸酸性磷酸酶阳性多核细胞比例显著升高(n=9,P<0.01),抗酒石酸酸性磷酸酶活性显著增强(n=9,P<0.0001),牛骨片上的骨吸收面积显著增大(n=8,P<0.0001),表明雌激素缺乏骨细胞分泌的可溶性因子可显著增强破骨生成与骨吸收功能;硬骨素抗体处理后,E组与ED组CM诱导的破骨生成与骨吸收均显著降低,其中E组抗酒石酸酸性磷酸酶阳性细胞比例降低(n=9,P<0.05),抗酒石酸酸性磷酸酶活性降低(n=9,P<0.0001),ED组抗酒石酸酸性磷酸酶阳性细胞比例降低(n=9,P<0.01),抗酒石酸酸性磷酸酶活性降低(n=9,P<0.0001),两组的骨吸收面积均显著降低(n=8,P<0.0001);双向方差分析显示,雌激素与硬骨素抗体对骨吸收的抑制作用存在显著交互效应(P=0.0002)。
对应实验结果图:
3.3 共培养实验验证骨细胞与破骨前体细胞的直接相互作用
实验目的:通过细胞-细胞直接接触共培养实验,验证雌激素缺乏骨细胞是否可通过直接接触信号增强破骨生成,以及硬骨素抗体的干预效果。
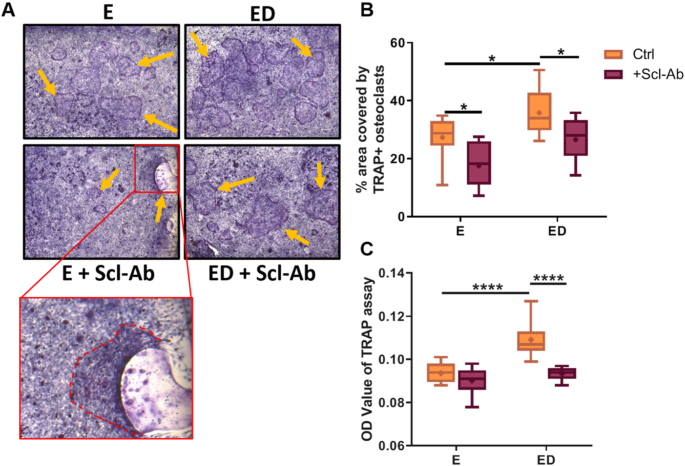
方法细节:将不同处理组的骨细胞接种于48孔板,24小时后将BMM或RAW264.7细胞接种于骨细胞上层,BMM接种密度为34000个/孔,RAW264.7细胞接种密度为10000个/孔;共培养体系中添加15ng/ml核因子κB受体活化因子配体,硬骨素抗体处理组全程添加300ng/ml硬骨素抗体以模拟体内全身给药方式;培养6天后进行抗酒石酸酸性磷酸酶染色,定量抗酒石酸酸性磷酸酶阳性细胞比例,同时检测培养上清中的抗酒石酸酸性磷酸酶活性。
结果解读:与E组骨细胞共培养的BMM相比,ED组骨细胞共培养的BMM中抗酒石酸酸性磷酸酶阳性细胞比例显著升高(n=9,P<0.05),抗酒石酸酸性磷酸酶活性显著增强(n=9,P<0.0001),表明雌激素缺乏骨细胞可通过直接接触信号增强破骨生成;硬骨素抗体处理后,E组共培养体系中抗酒石酸酸性磷酸酶阳性细胞比例降低(n=9,P<0.05),ED组共培养体系中抗酒石酸酸性磷酸酶阳性细胞比例与抗酒石酸酸性磷酸酶活性均显著降低(n=9,P<0.05和P<0.0001),且ED组的破骨生成水平恢复至与E组相当的水平,证实硬骨素抗体可逆转雌激素缺乏骨细胞通过直接接触介导的促破骨生成作用。
对应实验结果图:
3.4 分子水平解析破骨生成相关基因的表达变化
实验目的:从转录水平解析雌激素缺乏与硬骨素抗体处理对骨细胞中破骨生成调控基因、Wnt通路相关基因,以及破骨细胞中分化与功能相关基因表达的影响,明确硬骨素抗体的作用机制。
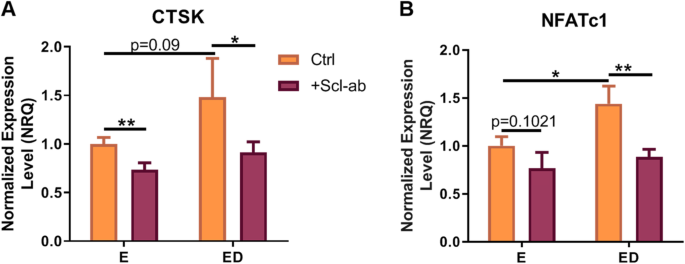
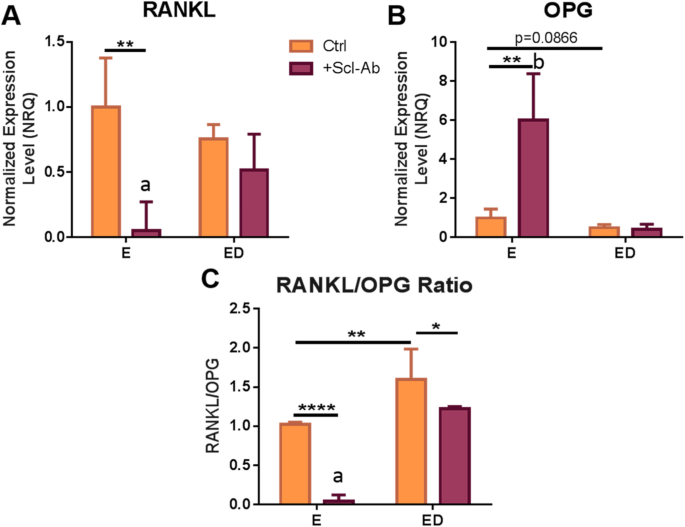
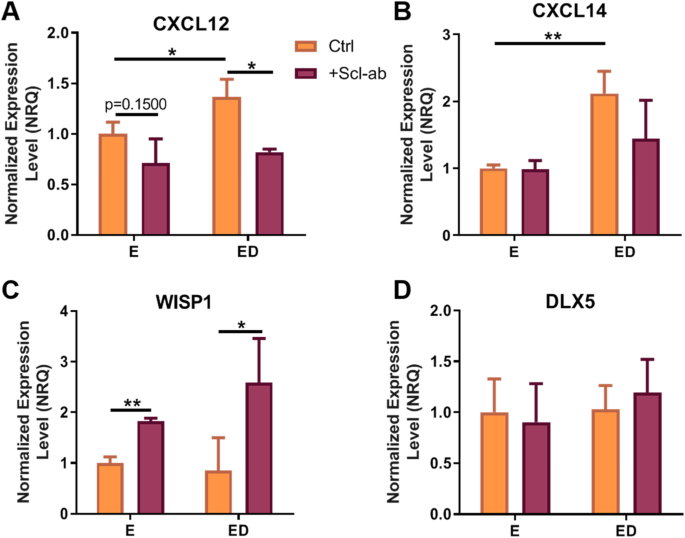
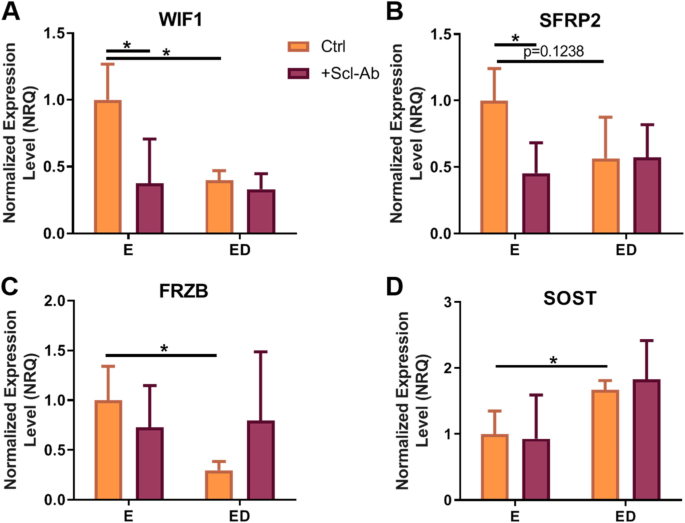
方法细节:骨细胞经机械刺激24小时后提取总RNA,采用qRT-PCR检测核因子κB受体活化因子配体、骨保护素、趋化因子(CXCL12、CXCL14)、破骨生成调控因子(WISP1、DLX5)及Wnt通路拮抗剂(WIF1、FRZB、SFRP2、SOST)的表达水平;BMM经不同组别的CM处理5天后提取总RNA,采用qRT-PCR检测破骨分化关键转录因子活化T细胞核因子胞浆1型(NFATc1)与骨吸收关键蛋白酶组织蛋白酶K(CTSK)的表达水平。
结果解读:骨细胞层面,ED组的核因子κB受体活化因子配体/骨保护素比值显著高于E组(n=9,P<0.01),硬骨素抗体处理后E组与ED组的核因子κB受体活化因子配体/骨保护素比值均显著降低(E组P<0.0001,ED组P<0.05);ED组中CXCL12与CXCL14的表达水平显著上调(n=9,P<0.05和P<0.01),硬骨素抗体处理后ED组CXCL12的表达水平显著下调(n=9,P<0.05);E组与ED组经硬骨素抗体处理后,WISP1的表达水平均显著上调(n=9,P<0.01和P=0.05);Wnt通路拮抗剂方面,ED组中WIF1、FRZB的表达水平显著下调(n=9,P<0.05),SOST的表达水平显著上调(n=9,P<0.05),硬骨素抗体处理对ED组的Wnt拮抗剂表达无显著影响,但可下调E组中WIF1与SFRP2的表达水平(n=9,P<0.05)。破骨细胞层面,ED组CM处理的BMM中活化T细胞核因子胞浆1型的表达水平显著上调(n=9,P<0.05),组织蛋白酶K表达水平呈上升趋势但无统计学显著性(n=9,P=0.09);硬骨素抗体处理后,E组与ED组CM处理的BMM中组织蛋白酶K的表达水平均显著下调(n=9,P<0.01和P<0.05),ED组CM处理的BMM中活化T细胞核因子胞浆1型的表达水平显著下调(n=9,P<0.01)。
对应实验结果图:
4. Biomarker研究及发现成果解析
本研究聚焦骨细胞中破骨生成调控相关的生物标志物,包括核因子κB受体活化因子配体/骨保护素比值、趋化因子CXCL12、破骨生成负调控因子WISP1,以及破骨细胞中的活化T细胞核因子胞浆1型、组织蛋白酶K,通过体外实验明确了这些生物标志物在雌激素缺乏状态下的表达变化及硬骨素抗体的调控作用,为绝经后骨质疏松症的诊断与治疗提供了潜在的靶点与机制依据。
本研究涉及的生物标志物可分为两类,一类是骨细胞来源的破骨生成调控标志物,包括核因子κB受体活化因子配体/骨保护素比值(经典骨重塑调控指标)、CXCL12(破骨前体细胞趋化因子)、WISP1(破骨生成负调控因子),筛选逻辑为基于骨细胞与破骨细胞间的已知信号通路,通过qRT-PCR检测不同处理组的表达差异;另一类是破骨细胞来源的分化与功能标志物,包括活化T细胞核因子胞浆1型(破骨分化关键转录因子)、组织蛋白酶K(骨吸收关键蛋白酶),筛选逻辑为基于破骨细胞分化与功能的核心分子,检测其在不同条件培养基处理后的表达变化。
骨细胞来源标志物中,核因子κB受体活化因子配体/骨保护素比值在雌激素缺乏组骨细胞中显著升高(n=9,P<0.01),硬骨素抗体处理后该比值在雌激素处理组与雌激素缺乏组均显著降低,其中雌激素处理组降低幅度更为显著(P<0.0001);CXCL12在雌激素缺乏组骨细胞中表达上调(n=9,P<0.05),硬骨素抗体处理后其表达水平显著下调(n=9,P<0.05);WISP1在硬骨素抗体处理后的雌激素处理组与雌激素缺乏组骨细胞中均显著上调(n=9,P<0.01和P=0.05)。破骨细胞来源标志物中,活化T细胞核因子胞浆1型在雌激素缺乏组条件培养基处理的破骨前体细胞中表达上调(n=9,P<0.05),硬骨素抗体处理后雌激素缺乏组条件培养基处理的破骨前体细胞中活化T细胞核因子胞浆1型表达显著下调(n=9,P<0.01);组织蛋白酶K在硬骨素抗体处理后的两组条件培养基处理的破骨前体细胞中均显著下调(n=9,P<0.01和P<0.05)。
本研究首次证实核因子κB受体活化因子配体/骨保护素比值、CXCL12可作为雌激素缺乏骨细胞促破骨生成的关键生物标志物,其中核因子κB受体活化因子配体/骨保护素比值的升高是雌激素缺乏骨细胞增强破骨生成的核心分子基础;同时,WISP1可作为硬骨素抗体干预的响应标志物,其表达上调是硬骨素抗体抑制破骨生成的重要机制之一。这些生物标志物的变化具有明确的统计学显著性,为绝经后骨质疏松症的发病机制研究提供了新的分子靶点,也为硬骨素抗体的临床疗效评估提供了潜在的细胞层面生物标志物。此外,本研究明确了硬骨素抗体可通过多靶点调控逆转雌激素缺乏骨细胞的促破骨生成作用,包括降低核因子κB受体活化因子配体/骨保护素比值、下调CXCL12、上调WISP1,为硬骨素抗体的临床应用提供了更全面的机制依据。