1. 领域背景与文献引入
文献英文标题:Prediction of acid radical ion binding residues by K-nearest neighbors classifier;发表期刊:BMC Molecular and Cell Biology;影响因子:未公开;研究领域:生物信息学-蛋白质配体结合预测
蛋白质是生命活动的核心执行者,其功能的发挥高度依赖于与特定配体的相互作用,酸根离子(如磷酸根、硫酸根、亚硝酸根等)是一类关键的蛋白质配体,参与酶活性磷酸化调控、半胱氨酸合成、蛋白质硫酸化等多种细胞代谢与功能调控过程。早期研究主要依赖实验方法定位酸根离子结合残基,虽能获得精准的位点信息,但存在通量低、成本高、难以大规模分析的局限性。随着生物信息学的发展,计算预测方法逐渐成为酸根离子结合残基研究的主流,2016年的IonSeq模型、2017年的支持向量机(SVM)算法等为该领域提供了高效工具,但现有方法在亚硝酸根、碳酸根等酸根离子的预测性能上仍有提升空间,尤其是缺乏对序列组成与位置保守性特征的高效整合与降维优化。
针对这一研究空白,本研究构建了基于K近邻(KNN)分类器的酸根离子结合残基预测模型,通过优化特征提取策略与模型参数,提升了多种酸根离子结合残基的预测性能,为蛋白质功能解析与药物分子设计提供了更精准的计算工具。
2. 文献综述解析
作者按研究方法的类型将领域现有研究分为实验研究与计算预测研究两类,系统梳理了不同方法的技术路径、核心结论及局限性,通过对比现有方法的性能短板,凸显了本研究在特征整合与模型优化上的创新价值。
实验研究方面,1966年Pardee等通过实验方法纯化了沙门氏菌中与硫酸根结合的蛋白质,定位了结合残基并解析了相互作用机制;2002年Richard等采用实验方法研究了蛋白聚糖与硫酸根的相互作用,精准定位了蛋白质中与硫酸乙酰肝素的结合位点;2003年Tamada等通过实验方法实现了丝素蛋白的硫酸化修饰并分析了其抗凝活性。实验方法的优势在于能直接验证结合残基的功能,结果可靠性高,但局限性是通量极低,仅能针对单个或少量蛋白质进行分析,难以满足蛋白质组层面的大规模研究需求。
计算预测研究方面,2016年Hu等开发的IonSeq模型,基于BioLip数据库构建数据集,实现了四种酸根离子结合残基的近98%准确率预测;同年Hu等采用集成分类器预测硫酸根与磷酸根结合残基,马修相关系数(MCC)高于0.23,整体准确率高于97%;2017年Li等采用SVM算法识别硫酸根结合残基,五折交叉验证的马修相关系数达0.571,整体准确率为78.5%;2018年Peyton等采用可解释的提升算法,基于配体化学亚结构与蛋白质序列基序预测蛋白质-配体相互作用,获得了较高的准确率。计算方法的优势在于通量高、成本低,能快速处理大规模数据,但局限性是不同方法在不同酸根离子的预测性能上差异较大,尤其是亚硝酸根、碳酸根的预测性能仍有明显提升空间,且现有方法对序列组成与位置保守性的特征整合不足,存在特征冗余导致的模型泛化能力弱的问题。
与现有研究相比,本研究的创新点在于首次采用增量多样性(ID)算法对组成特征进行降维,同时整合位置保守性特征构建20维组合特征,采用K近邻分类器提升了亚硝酸根、碳酸根等酸根离子结合残基的预测性能,其中亚硝酸根的马修相关系数从现有方法的约0.23提升至0.612,有效弥补了现有方法在这些离子预测上的不足。
3. 研究思路总结与详细解析
本文的研究目标是构建精准的酸根离子结合残基预测模型,核心科学问题是如何通过优化特征参数与模型参数,提升K近邻分类器对酸根离子结合残基的识别能力,技术路线遵循“数据集构建→特征提取与优化→模型参数调优→多维度验证”的闭环逻辑,确保模型的准确性与通用性。
3.1 数据集构建与预处理
实验目的:构建无冗余、高质量的酸根离子结合残基数据集,为模型训练与验证提供可靠的样本基础。
方法细节:从BioLip数据库下载13种酸根离子的蛋白质相互作用原始数据,首先筛选出分辨率小于3Å、序列长度大于50残基的蛋白质,再采用CD-HIT软件去除序列一致性高于30%的冗余蛋白质,最终保留亚硝酸根(NO₂⁻)、碳酸根(CO₃²⁻)、硫酸根(SO₄²⁻)、磷酸根(PO₄³⁻)四种酸根离子的结合残基数据;采用滑动窗口法,分别设置窗口长度为5、7、9、11、13、15、17,将蛋白质序列切割为重叠片段,若片段中心残基为实验验证的酸根离子结合残基则归为正样本,否则归为负样本,为保证每个残基都能作为中心残基,在蛋白质序列两端添加(窗口长度-1)/2个虚拟残基“X”。
结果解读:经预处理后的数据集去除了冗余信息,保证了样本的多样性与可靠性,四种酸根离子的样本量满足统计分析要求,为后续特征提取与模型训练提供了标准化输入。
产品关联:文献未提及具体实验产品,领域常规使用BioLip数据库获取蛋白质-配体相互作用数据,CD-HIT软件进行序列冗余去除,Python或R语言进行数据预处理。
3.2 特征参数提取与精炼
实验目的:提取能有效区分结合残基与非结合残基的特征参数,通过降维处理减少特征冗余,提升模型的预测效率与泛化能力。
方法细节:提取五类基础特征,包括氨基酸组成、极化电荷组成(分为正电荷、负电荷、不带电荷三类)、亲水-疏水组成(分为六类)、二级结构组成(分为α-螺旋、β-折叠、无规卷曲三类)、相对溶剂可及性组成(以25%为阈值分为暴露与埋藏两类);采用位置权重评分矩阵(PWSM)提取上述五类特征的位置保守性信息,构建位置组合特征;采用增量多样性(ID)算法对组成特征进行降维,将20维氨基酸组成压缩为2维ID值,同理对极化电荷、亲水-疏水等组成特征进行降维,最终得到10维ID值;对位置特征进行降维处理,得到10维位置评分值,将两类降维特征整合为20维组合特征。
结果解读:降维后的组合特征保留了组成与位置的关键信息,有效减少了特征冗余,与单一的组成特征或位置特征相比,能更精准地识别酸根离子结合残基。
产品关联:文献未提及具体实验产品,领域常规使用PSIPRED等工具预测蛋白质二级结构,使用Python的scikit-learn库进行特征提取与降维。
3.3 模型参数优化与性能评估
实验目的:确定K近邻分类器的最优k值与窗口长度,评估不同特征参数的预测性能,验证模型的准确性与通用性。
方法细节:采用五折交叉验证评估模型性能,将数据集随机分为5份,4份作为训练集,1份作为测试集,重复5次取平均值;为解决样本不平衡问题,每次实验随机采样与正样本数量相等的负样本,重复10次取平均值;针对每个窗口长度与特征参数,通过测试不同k值对应的马修相关系数(MCC)确定最优k值;分别测试组成组合特征、位置组合特征、20维组合特征的预测性能,评估指标包括敏感性(Sn)、特异性(Sp)、准确率(Acc)、马修相关系数(MCC)、假阳性率(FPR);同时进行独立测试,将数据集按8:2分为训练集与测试集,评估模型的泛化能力;此外,用该模型预测六种金属离子(Zn²⁺、Fe²⁺、Fe³⁺、Cu²⁺、Mn²⁺、Co²⁺)的结合残基,验证方法的通用性。
结果解读:五折交叉验证结果显示,20维组合特征的预测性能最优,四种酸根离子的Acc、Sn均高于70%,MCC均高于0.45,FPR低于30.8%;其中亚硝酸根的MCC达0.612,Acc达80.6%,Sn与Sp分别为81.6%与79.6%,性能提升最为明显;独立测试结果显示,Sn高于40.9%,Acc与Sp高于84.2%,MCC高于0.116,FPR低于15.4%;与IonSeq模型相比,本文模型的Sn更高,在NO₂⁻、CO₃²⁻的MCC上表现更优;预测金属离子结合残基的结果显示,Acc、Sn、Sp均高于77.6%,MCC高于0.6,FPR低于19.6%,验证了方法的通用性。
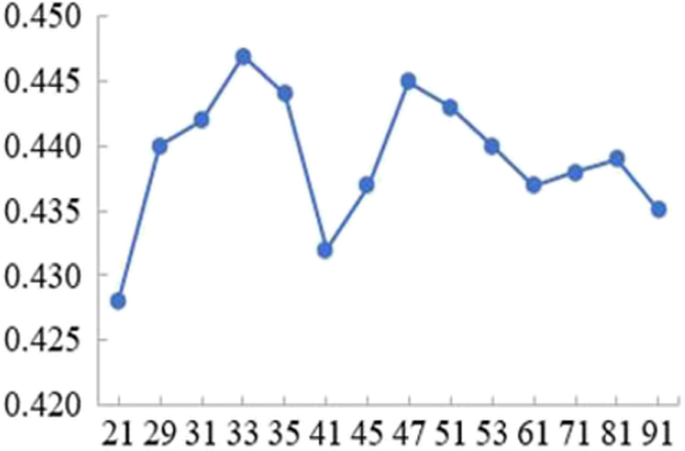
图1展示了硫酸根离子在窗口长度13时,k值与MCC的关系,当k=33时MCC达到最高值0.447,为该特征下的最优k值,通过10次重复实验确定平均最优k值为33。
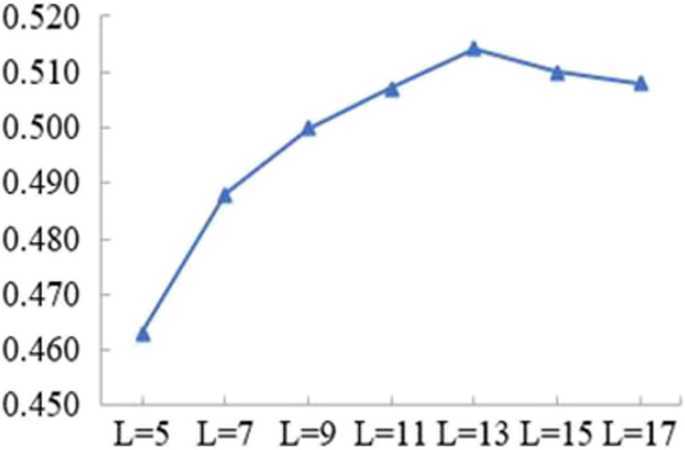
图2展示了磷酸根离子在不同窗口长度下的MCC值,窗口长度从5增加到13时MCC逐渐升高,从13增加到17时MCC逐渐降低,确定窗口长度13为最优窗口长度,此时MCC达最高值。
产品关联:文献未提及具体实验产品,领域常规使用WEKA平台实现K近邻分类器,采用五折交叉验证与独立测试进行模型评估。
4. Biomarker研究及发现成果解析
本文涉及的Biomarker为酸根离子结合残基,属于蛋白质功能位点Biomarker,通过“数据库实验数据筛选→计算模型预测→多维度验证”的逻辑进行识别与验证,为蛋白质功能解析与药物靶点发现提供了高效工具。
本研究的Biomarker包括亚硝酸根(NO₂⁻)、碳酸根(CO₃²⁻)、硫酸根(SO₄²⁻)、磷酸根(PO₄³⁻)四种酸根离子的蛋白质结合残基,筛选与验证逻辑为:从BioLip数据库获取实验验证的蛋白质-酸根离子相互作用数据,经冗余去除后构建数据集,通过计算模型预测潜在的结合残基,再经五折交叉验证、独立测试及金属离子结合残基预测验证方法的可靠性。
Biomarker的来源为BioLip数据库中的实验验证蛋白质-酸根离子相互作用数据,涵盖了不同物种、不同功能的蛋白质;验证方法包括五折交叉验证与独立测试,特异性与敏感性数据显示,五折交叉验证中20维组合特征的敏感性(Sn)高于70%,特异性(Sp)高于69.2%,马修相关系数(MCC)高于0.45;独立测试中Sn高于40.9%,Sp高于84.2%,MCC高于0.116;预测金属离子结合残基的结果显示,Sn高于77.6%,Sp高于77.6%,MCC高于0.6,进一步验证了方法的特异性与通用性。
该Biomarker的功能关联为参与蛋白质活性调控、细胞代谢等关键生命过程,例如磷酸根结合残基参与酶的磷酸化修饰,调控酶活性;硫酸根结合残基参与半胱氨酸合成与蛋白质硫酸化过程。本研究的创新性在于首次采用结合组成与位置保守性的降维特征,通过K近邻分类器提升了NO₂⁻、CO₃²⁻等酸根离子结合残基的预测性能,其中NO₂⁻的MCC达0.612(五折交叉验证,文献未明确样本量),CO₃²⁻的假阳性率降至21.5%(五折交叉验证,文献未明确样本量);预测金属离子结合残基的结果显示,MCC高于0.6,为蛋白质功能研究提供了高效的计算工具,同时为药物设计中靶点蛋白的配体结合位点预测提供了技术支撑。