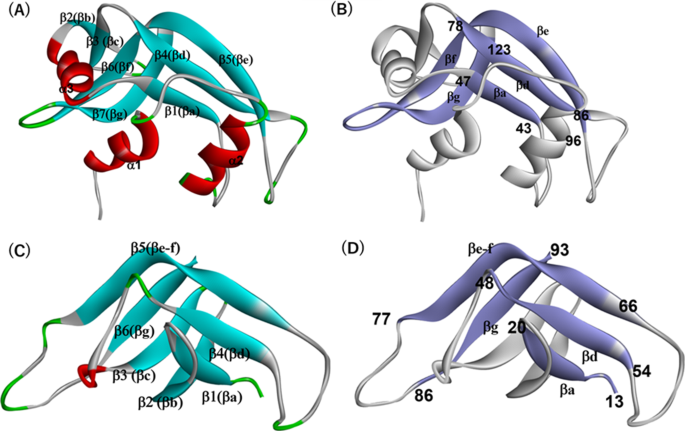
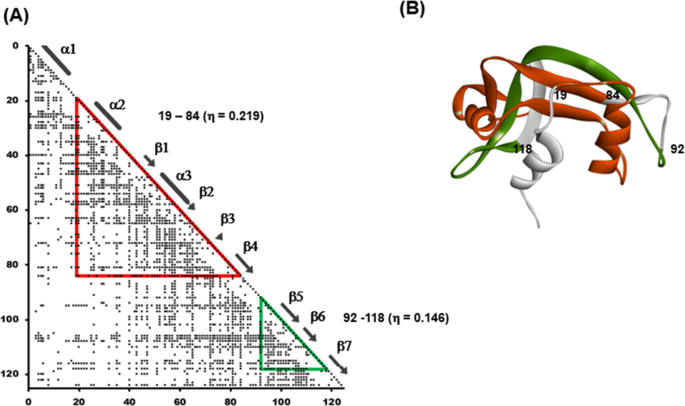
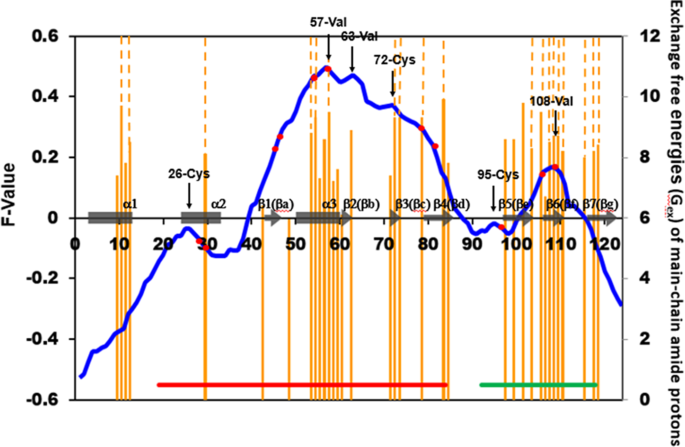
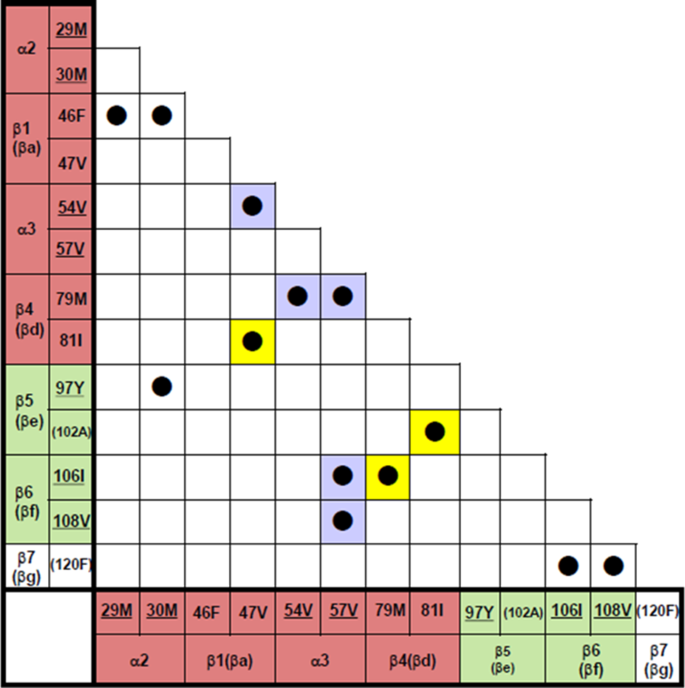
Abstract
BACKGROUND: Protein folding is a complex process in which amino acid sequences encode the information required for a polypeptide chain to fold into its functional three-dimensional (3D) structure. Many proteins share common substructures and recurring secondary structure elements that contribute to similar 3D folding patterns, even across different protein families. This study examines two distinct groups of proteins, the RNase A-like fold and the trypsin-like serine protease fold, classified by SCOPe. These proteins share only some substructures that contribute to their folding cores. Despite minimal sequence identity, they exhibit partial structural similarities in their 3D topologies. We used a sequence-based approach, including inter-residue average distance statistics and contact frequency prediction, to explore these folding characteristics. Structural observations guided further analyses of conserved hydrophobic residue packing, highlighting key folding units within each fold. RESULTS: Our analysis predicted two compact regions within each protein group. Interactions between these regions form a partially shared topology. We identified conserved hydrophobic residues critical to these interactions, suggesting a common mechanism for establishing these structural features. Despite overall structural differences between the RNase A-like and trypsin-like folds, our findings emphasize the presence of a shared partial folding core. CONCLUSIONS: The partially shared structural features in the RNase A-like and trypsin-like serine protease folds reflect a convergent folding mechanism. This mechanism underscores the evolutionary adaptation of protein folding, where distinct folds can still retain critical, conserved structural motifs. These findings highlight how proteins with overall different topologies can evolve to share key folding features, demonstrating the elegance and efficiency of protein evolution.