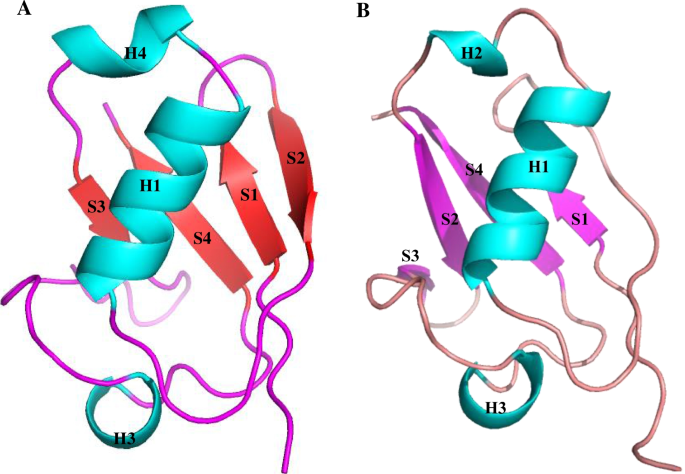
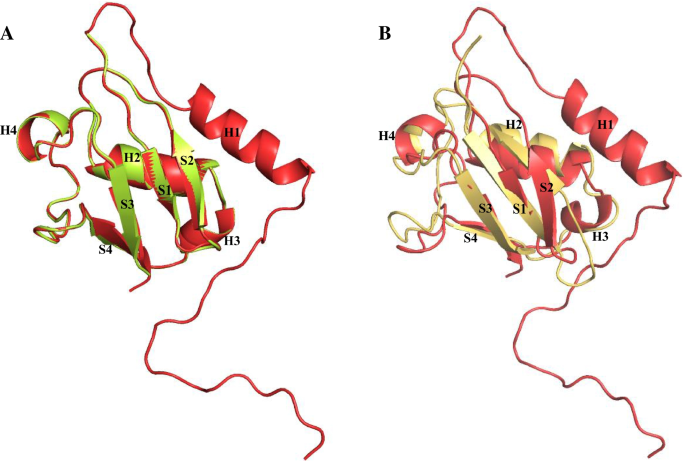
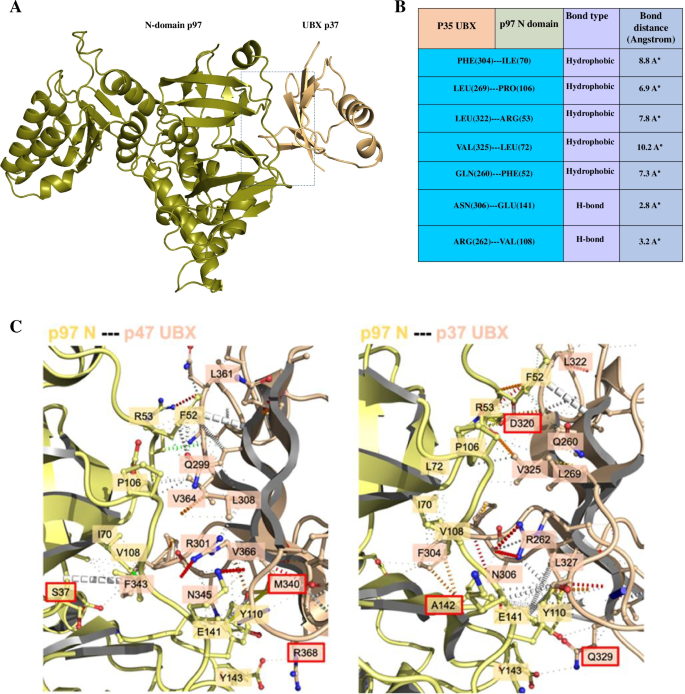
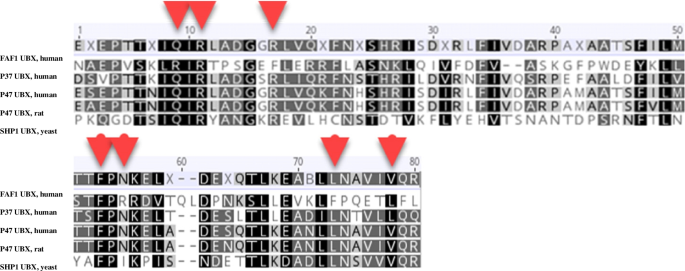
Abstract
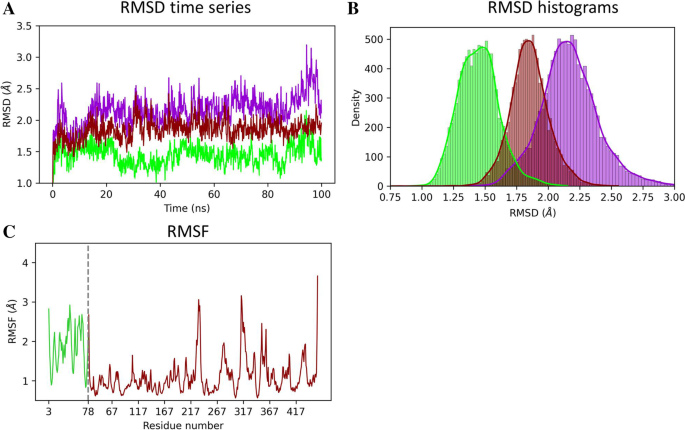
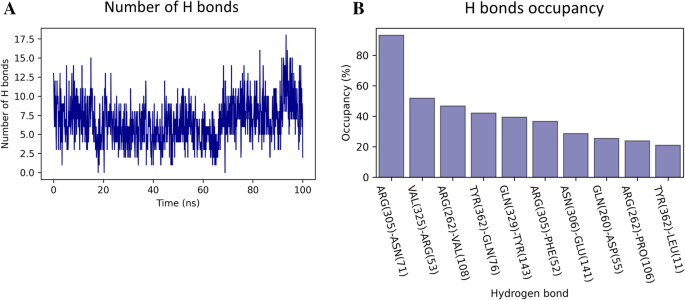
BACKGROUND: The AAA + ATPase p97 is an essential unfoldase/segragase involved in a multitude of cellular processes. It functions as a molecular machine critical for protein homeostasis, homotypic membrane fusion events and organelle biogenesis during mitosis in which it acts in concert with cofactors p47 and p37. Cofactors assist p97 in extracting and unfolding protein substrates through ATP hydrolysis. In contrast to other p97's cofactors, p37 uniquely increases the ATPase activity of p97. Disease-causing mutations in p97, including mutations that cause neurodegenerative diseases, increase cofactor association with its N-domain, ATPase activity and improper substrate processing. Upregulation of p97 has also been observed in various cancers. This study aims towards the characterization of the protein-protein interaction between p97 and p37 at the atomic level. We defined the interacting residues in p97 and p37. The knowledge will facilitate the design of unique small molecules inhibiting this interaction with insights into cancer therapy and drug design. RESULTS: The homology model of human p37 UBX domain was built from the X-ray crystal structure of p47 C-terminus from rat (PDB code:1S3S, G) as a template and assessed by model validation analysis. According to the HDOCK, HAWKDOCK, MM-GBSA binding free energy calculations and Arpeggio, we found that there are several hydrophobic and two hydrogen-bonding interactions between p37 UBX and p97 N-D1 domain. Residues of p37 UBX predicted to be involved in the interactions with p97 N-D1 domain interface are highly conserved among UBX cofactors. CONCLUSION: This study provides a reliable structural insight into the p37-p97 complex binding sites at the atomic level though molecular docking coupled with molecular dynamics simulation. This can guide the rational design of small molecule drugs for inhibiting mutant p97 activity.