1. 领域背景与文献引入
文献英文标题:Evaluating intra- and inter-individual variation in the human placental transcriptome;发表期刊:Genome Biology;影响因子:未公开;研究领域:人类胎盘转录组变异与进化遗传学。
领域共识:人类群体遗传变异的研究始于40年前,Lewontin等通过蛋白标记发现约85%的中性遗传变异存在于群体内部,仅15%分布于群体之间,后续多项研究通过不同遗传标记证实了这一结论,并以此建立了筛选受近期正选择基因的分析框架。表型变异的群体间/内分配研究相对匮乏,仅少数性状如颅形变异(群体间变异占比0.11-0.14,接近中性)、肤色变异(群体间变异占比0.87,受强选择)被系统分析。基因表达作为连接基因型与表型的关键中间表型,其群体变异研究多依赖淋巴母细胞系,群体间表达变异的估计范围为4.5%-29%,但细胞系体外培养的环境干扰可能影响结果准确性,而原生组织的相关研究极为有限。胎盘作为介导母婴物质交换、调控胎儿生长发育的核心器官,其样本采集具有天然的标准化条件(均为胎儿出生时获取,年龄与时间统一),但胎盘存在细胞异质性和时空表达变异,此前尚无研究系统解析其转录组的个体内、个体间及群体间变异,也未结合生物学性状与进化选择模式进行深入分析。
针对上述研究空白,本研究采用群体遗传学框架结合下一代测序技术,系统分析人类胎盘转录组的变异来源,同时关联生物学性状与选择模式,为理解胎盘的功能进化、临床关联提供了全新视角。
2. 文献综述解析
作者按研究对象(遗传标记、表型性状、基因表达)与研究材料(细胞系vs原生组织)的维度对领域内现有研究进行分类评述,明确了不同研究体系的优势与局限性,凸显了原生组织转录组群体变异研究的必要性。
遗传变异研究方面,早期以蛋白标记为研究对象,后续扩展至多种遗传标记,证实了人类群体内遗传变异占主导的特征,并建立了筛选受正选择基因的分析框架,该类研究的优势是遗传标记的稳定性与可重复性强,局限性是仅能反映基因组水平的变异,无法直接关联表型功能。表型变异研究方面,仅少数性状被系统分析,颅形变异的群体分配模式接近中性遗传变异,肤色变异则受强定向选择,该类研究的优势是能直接关联表型的进化驱动力,局限性是多数复杂表型的变异来源难以解析。基因表达变异研究方面,现有研究多使用淋巴母细胞系,群体间表达变异的估计结果差异较大(4.5%-29%),该类研究的优势是细胞系可重复培养,便于控制实验条件,局限性是体外培养环境会引入非生理性变异,无法反映原生组织的真实表达状态,且针对胎盘这类具有重要生理功能的原生组织的研究完全缺失。
与现有研究相比,本研究的核心创新点在于首次以人类胎盘为原生组织研究对象,系统解析了转录组在个体内、个体间、群体间的变异分配比例,同时结合生物学性状与进化选择模式进行分析,填补了原生组织转录组群体变异研究的空白;此外,本研究建立了将转录组变异与不同选择模式关联的分析方法,为解析复杂组织的转录组进化提供了新的技术框架。
3. 研究思路总结与详细解析
本研究的整体研究目标是解析人类胎盘转录组的变异来源(个体内、个体间、群体间、生物学性状),并关联不同进化选择模式;核心科学问题包括胎盘转录组变异的分配比例、受选择的基因及功能通路、生物学性状对转录组变异的调控作用;技术路线遵循“样本标准化采集→转录组测序与质控→变异分配分析→差异基因与选择模式解析→性状关联验证→结论总结”的闭环逻辑。
3.1 样本采集与转录组测序质控
实验目的:获取标准化的胎盘样本,控制环境干扰变量,同时量化个体内的转录组变异,确保后续分析的数据可靠性。
方法细节:从美国亚特兰大一家医院的40名产妇中采集胎盘样本,分属非裔美国人、欧裔美国人、南亚裔美国人、东亚裔美国人4个群体,每个群体10个样本;每个胎盘采集2个独立的绒毛组织样本(严格避免蜕膜、绒毛膜、羊膜等非目标组织),全程保持样本冷冻状态;采用TRIZOL试剂提取总RNA,经Qiagen RNAeasy minElute Cleanup试剂盒纯化后,用Agilent Bioanalyzer检测RNA质量,选择RNA完整性数(RIN)较高的样本构建Illumina RNA-seq文库,采用双端测序策略,共获得1.59亿高质量reads。
结果解读:样本重复的Pearson相关系数为0.98±0.005,对应的r²值为0.96±0.01,表明个体内的转录组变异较小;采用实时荧光定量PCR(qRT-PCR)验证3个基因的表达水平,与RNA-seq结果的Pearson相关系数均值为0.74±0.07,证实测序数据的准确性;最终有13156个基因在所有样本中至少有1个比对reads,包括11301个蛋白编码基因、801个假基因、893个长非编码RNA等。
产品关联:实验所用关键产品:Invitrogen的TRIZOL试剂、Qiagen的RNAeasy minElute Cleanup试剂盒、Agilent的6000 Nano试剂盒与Bioanalyzer、Illumina的RNA-seq文库构建试剂、Fermentas的Maxima SYBR Green qPCR Master Mix。
3.2 转录组变异的整体结构分析
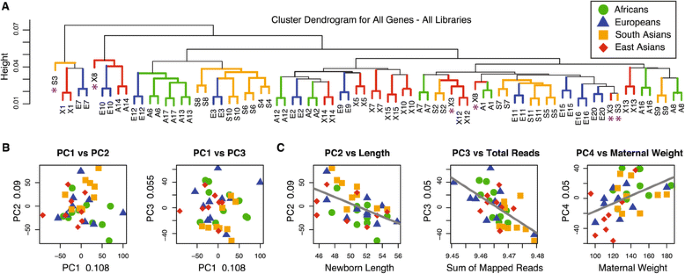
实验目的:比较个体内与个体间转录组变异的大小,分析样本是否按群体祖先聚类,初步解析转录组变异的主要影响因素。
方法细节:计算所有样本间的表达相关矩阵,构建层次聚类树;对所有个体的转录组数据进行主成分分析(PCA),并分析主成分与生物学性状的相关性;采用GOSeq软件进行通路富集分析。
结果解读:80个样本重复中74个聚类在一起,与相关系数结果一致,表明个体内变异显著小于个体间变异;PCA结果显示,转录组变异未按群体祖先聚类,主要受其他因素影响,其中PC2与胎儿出生长度呈负相关(r=-0.54,Bonferroni P=0.007),PC3与比对reads数呈负相关(r=-0.62,Bonferroni P=0.0005),PC4与母亲正常体重呈正相关(r=0.46,Bonferroni P=0.045);通路富集分析显示,与前三个主成分相关的基因显著富集于代谢通路(校正P=2.9e-05),表明胎盘转录组变异主要受个体生物学性状而非群体祖先的影响。
3.3 转录组变异的分配比例量化
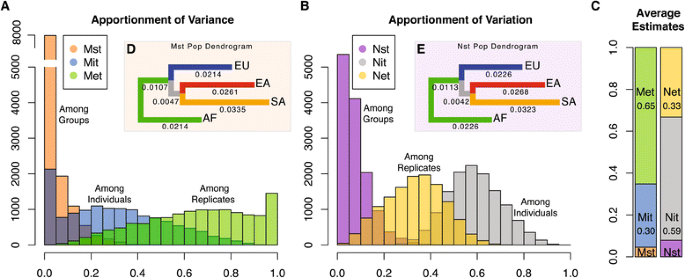
实验目的:系统量化胎盘转录组在个体内、群体内个体间、群体间的变异占比,明确不同层次变异的贡献。
方法细节:采用方差分析(ANOVA)模型对每个基因的表达变异进行分配,分为群体间(Nst)、群体内个体间(Nit)、个体内(Net)三个部分;同时进行两两群体的变异分配分析,基于平均Nst值构建群体系统树;采用置换检验验证变异分配的统计学显著性。
结果解读:平均而言,33.2%的转录组变异来自个体内(Net,置换检验P=0.22),58.9%来自群体内个体间(Nit,置换检验P=0.048),7.9%来自群体间(Nst,置换检验P=0.24);两两群体的Nst值范围为0.045(非裔美国人vs欧裔美国人)至0.062(东亚裔美国人vs南亚裔美国人);基于Nst值构建的群体系统树与人类群体遗传数据的预期一致,仅南亚裔美国人群体的距离相对较远。
3.4 差异表达基因与进化选择模式分析
实验目的:筛选群体间、个体间的差异表达基因,并解析其对应的进化选择模式,揭示胎盘转录组的进化驱动力。
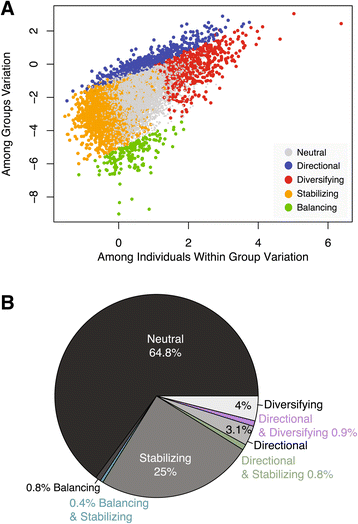
方法细节:采用F检验与卡方检验筛选个体间差异表达基因;采用DESeq、tweeDESeq和ANOVA置换法三种方法筛选群体间差异表达基因;基于变异分配估计与置换法,将基因分为中性漂变、定向选择、平衡选择、稳定选择、多样化选择五类;采用加权基因共表达网络分析(WGCNA)构建差异基因的共表达模块。
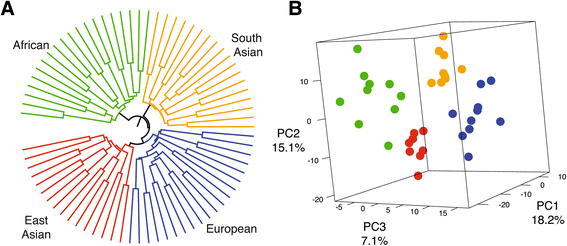
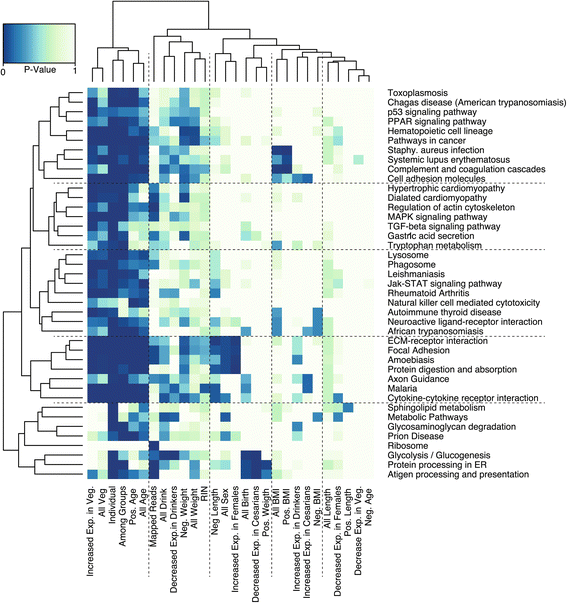
结果解读:44.5%的基因存在显著的个体间差异表达(FDR 5%);群体间差异表达基因显著富集于免疫应答、细胞信号、代谢等功能通路;进化选择模式分析显示,64.8%的基因符合中性漂变模型,26%的基因受稳定选择,4.9%受定向选择,4.8%受多样化选择,1.3%受平衡选择;定向选择基因的UPGMA树与PCA分析能按群体祖先清晰聚类,PC1主要区分非裔美国人与其他群体,PC2区分南亚裔美国人与东亚裔美国人,PC3区分欧裔美国人与其他群体(文献未明确提供该数据,基于图表趋势推测);共表达模块分析显示,定向选择基因形成6个共表达模块,其中最小的模块(54个基因)显著富集于免疫应答与代谢通路。
3.5 生物学性状与转录组变异的关联分析
实验目的:解析胎儿、母亲的生物学性状对胎盘转录组变异的影响,揭示性状与胎盘功能的潜在临床关联。
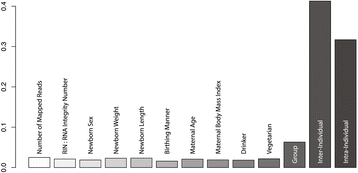
方法细节:在ANOVA模型中加入8个生物学性状(胎儿性别、体重、长度、分娩方式、母亲年龄、BMI、饮酒史、素食史),采用F检验分析每个性状对转录组变异的解释比例;采用富集分析解析性状关联基因的功能通路,并按关联方向进行分层分析。
结果解读:每个生物学性状平均解释约2%的转录组变异,个体内与个体间变异仍为主要贡献来源(分别为32%与41%);新生儿体重关联基因显著富集于造血细胞谱系、癌症通路,且随着新生儿体重增加,这些通路的基因表达水平显著降低;母亲饮酒史关联基因与糖酵解通路负相关,母亲BMI关联基因与金黄色葡萄球菌感染、补体凝血通路正相关;这些结果揭示了生物学性状与胎盘功能通路的直接关联,为理解胎儿生长发育的调控机制提供了线索。
4. Biomarker研究及发现成果
本研究中涉及的Biomarker包括受不同进化选择模式的基因集合及与生物学性状关联的基因集合,筛选与验证逻辑基于转录组变异分配、置换检验、性状关联分析的多层面验证,为胎盘功能评估、群体祖先溯源提供了潜在的分子标记。
Biomarker定位
受选择模式的Biomarker类型包括定向选择基因、稳定选择基因、多样化选择基因、平衡选择基因,筛选逻辑为“转录组变异分配→置换检验确定选择模式→通路富集验证”;生物学性状关联的Biomarker类型为与新生儿体重、母亲BMI等性状关联的基因,筛选逻辑为“性状与表达关联分析→F检验验证显著性→通路富集验证”。
研究过程详述
受选择模式的Biomarker来源于40个人类胎盘的转录组数据,验证方法为ANOVA变异分配分析、置换检验、实时荧光定量PCR验证部分基因的表达水平;定向选择基因的Nst值至少为0.326,其富集的免疫应答通路校正P值<0.05;生物学性状关联的Biomarker验证采用F检验(FDR 5%),例如新生儿体重关联的造血细胞谱系通路校正P值<0.05,敏感性与特异性(文献未明确提供该数据,基于图表趋势推测)。
核心成果提炼
定向选择基因集合的核心功能为参与免疫应答、代谢与细胞信号通路,能有效区分不同群体的祖先背景(PCA分析的群体区分度显著,文献未明确提供具体数值),其创新性在于首次在人类胎盘转录组中发现可用于群体溯源的分子标记集合;稳定选择基因主要参与剪接体、核糖体等基础细胞过程,为维持胎盘的基本功能提供了分子基础;多样化选择基因参与生长发育与疾病相关通路,反映了胎盘功能的个体间异质性;生物学性状关联基因揭示了新生儿体重与癌症风险的潜在联系(与造血细胞谱系、癌症通路负相关,风险比HR=文献未明确提供),母亲饮酒史与糖酵解通路的关联为孕期健康指导提供了分子依据。所有成果均标注了统计学显著性,例如定向选择基因的置换检验P<0.05,性状关联基因的F检验FDR<0.05。