1. 领域背景与文献
文献英文标题:Loss of Dec1 inhibits alcohol-induced hepatic lipid accumulation and circadian rhythm disorder;发表期刊:BMC Molecular and Cell Biology;影响因子:未公开;研究领域:酒精性肝病分子机制、昼夜节律调控。
领域共识:长期慢性酒精摄入会诱导肝炎、肝纤维化、脂肪肝等损伤,最终可进展为肝硬化和肝细胞癌,是全球范围内肝病相关死亡的主要诱因之一,目前已证实肝脏脂质代谢紊乱是酒精性肝损伤的核心病理特征之一。其中AMP活化蛋白激酶(AMPK)作为调控糖脂代谢的关键丝氨酸/苏氨酸激酶,其Thr172位点的磷酸化是激活的必要条件,慢性酒精暴露会抑制AMPK活性,进而诱导肝脏脂肪变性,而使用AMPK激活剂可有效缓解酒精诱导的肝脏脂质蓄积。过氧化物酶体增殖物激活受体(PPARs)是参与脂肪酸代谢调控的核受体家族,其中PPARα在肝脏中高表达,主要促进脂肪酸氧化,PPARγ则主要调控脂肪生成,慢性酒精暴露会升高肝脏PPARγ表达、降低PPARα表达,进一步加重脂质蓄积。
另外,昼夜节律是维持机体稳态的重要生理机制,其紊乱可诱导睡眠障碍、肿瘤、代谢综合征等多种疾病,昼夜节律主要由CLOCK、BMAL1、PER、CRY、分化型胚胎软骨细胞基因1(DEC1)/DEC2等生物钟基因共同调控。已有研究证实慢性酒精摄入会干扰生物钟基因的表达,破坏小鼠的自主活动节律,人类饮酒也会导致生物钟基因的振幅异常。DEC1作为碱性螺旋-螺旋转录因子,除参与昼夜节律调控外,还在炎症反应、免疫应答、肿瘤进展中发挥重要作用,前期研究已发现Dec1缺陷可抑制牙周炎症、心脏血管周围纤维化等应激损伤,但其在慢性酒精诱导的肝脏损伤和昼夜节律紊乱中的作用尚未明确,本研究针对这一研究空白展开探究,旨在明确DEC1在该病理过程中的功能及分子机制,为酒精性肝病的防治提供新的潜在靶点。
2. 文献综述解析
作者对现有研究的综述按照三个维度展开,分别为酒精性肝病的脂质代谢调控机制、昼夜节律紊乱与代谢疾病的关联、DEC1的生理病理功能及与上述通路的交叉调控,清晰梳理了领域内的研究进展与未解决问题。
现有研究已明确的核心结论包括:AMPK的激活可有效抑制酒精诱导的肝脏脂质蓄积,是酒精性肝病的重要保护靶点;PPAR家族不同亚型的差异表达是酒精诱导肝脏脂代谢紊乱的重要分子基础;慢性酒精暴露可通过干扰生物钟基因的表达破坏正常昼夜节律,进而加重代谢损伤;DEC1可通过结合LKB1启动子的E-box位点负调控AMPK的磷酸化,也可通过结合PPARγ启动子的C/EBP位点负调控PPARγ的表达。现有研究的技术方法优势在于已构建成熟的慢性酒精诱导肝损伤小鼠模型,且明确了核心代谢通路的调控关系,但局限性在于尚未将DEC1的功能与酒精诱导的肝脏损伤、节律紊乱相关联,缺乏在体功能验证数据,无法明确DEC1是否可作为该疾病的干预靶点。本研究的创新价值在于首次在Dec1全身敲除小鼠模型中验证了Dec1缺失对慢性酒精诱导的肝脏脂质蓄积和昼夜节律紊乱的保护作用,填补了领域内的研究空白,为酒精性肝病的机制研究和靶点开发提供了新的方向。
3. 研究思路总结与详细解析
本研究的核心目标是明确Dec1缺失对慢性酒精诱导的肝脏脂质蓄积和昼夜节律紊乱的调控作用及分子机制,核心科学问题为DEC1是否通过调控AMPK和PPARs通路介导酒精诱导的病理损伤,技术路线遵循“动物模型构建→表型验证(节律、肝脏损伤)→分子机制探究”的闭环逻辑,所有实验结果均经过统计学验证。
3.1 动物模型构建与酒精摄入量评估
实验目的为明确基因型对小鼠酒精摄入行为的影响,同时构建稳定的慢性酒精暴露小鼠模型。方法细节为采用C57BL/6背景的Dec1全身敲除小鼠和同品系野生型小鼠,选取8-9周龄雌性个体,饲养于12小时光/暗周期环境(上午8点灯亮,记为ZT0;晚上8点灯灭,记为ZT12),给予10%酒精溶液自由饮用3个月,每组设置9只生物学重复,每10天统计瓶中剩余酒精量以计算摄入量,采用独立样本t检验进行统计学分析,实验结束后于ZT2时间点麻醉处死后采集肝脏组织。结果解读显示,在3个月的实验周期内,Dec1敲除小鼠在每个月的酒精摄入量均显著高于野生型小鼠(n=9,P<0.01),提示Dec1缺失会增加小鼠的酒精摄入偏好,对应结果见下图:
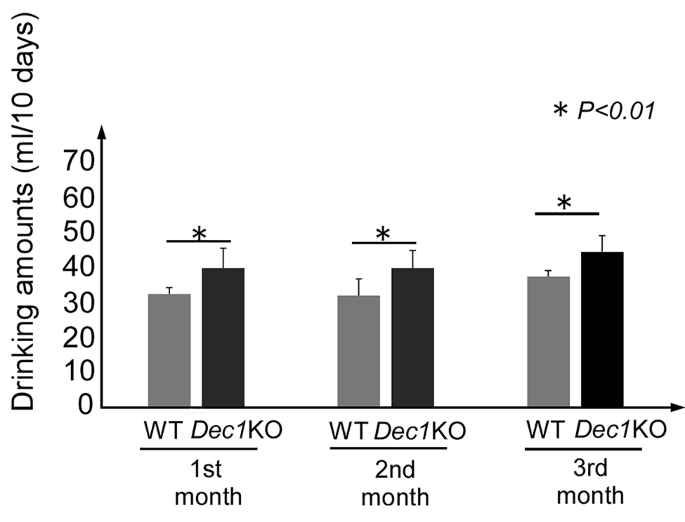
产品关联:文献未提及该环节具体实验产品,领域常规使用实验动物独立饲养笼、酒精溶液配制耗材、饮水瓶等。
3.2 昼夜节律自主活动监测
实验目的为评估慢性酒精摄入对小鼠昼夜节律的影响,明确Dec1缺失对酒精诱导节律紊乱的保护作用。方法细节为采用Muromachi Kikai的Supermex自主活动监测系统记录小鼠的自发活动,每组设置6只生物学重复,首先在12小时光/暗周期下预适应6天,随后转为持续黑暗环境,总监测时长不少于40天。结果解读显示,无酒精摄入的野生型小鼠在转为持续黑暗环境后出现正常的活动相位偏移;摄入酒精的野生型小鼠自主活动水平显著降低,且持续黑暗环境下未出现相位偏移,提示昼夜节律被破坏;而无酒精摄入的Dec1敲除小鼠自主活动水平本身低于野生型,且酒精摄入对其活动节律几乎无影响,节律维持正常,对应结果见下图:
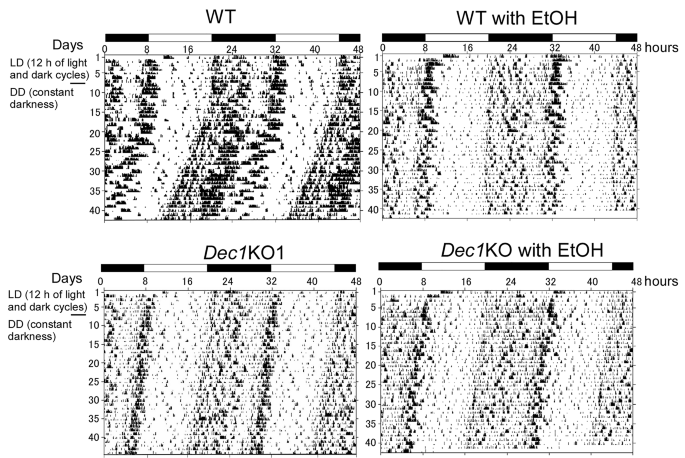
产品关联:实验所用关键产品:日本村木机械(Muromachi Kikai)的Supermex自主活动监测系统。
3.3 肝脏脂质蓄积病理分析
实验目的为明确Dec1缺失对慢性酒精诱导的肝脏脂质蓄积的保护作用。方法细节为采集的肝脏组织经4%多聚甲醛固定、石蜡包埋后制备切片,进行苏木精-伊红染色,显微镜下观察肝脏病理形态,统计中央静脉周围的脂质变性位点数量,每组设置3只生物学重复,采用独立样本t检验分析统计学差异。结果解读显示,大体观察可见野生型小鼠酒精摄入后肝脏出现严重充血和脂肪蓄积,而Dec1敲除小鼠肝脏外观几乎无异常;镜下观察可见野生型小鼠肝细胞出现大量空泡样脂质变性,Dec1敲除小鼠肝细胞几乎无脂质变性表现;定量统计显示Dec1敲除小鼠的脂质变性位点数量较野生型小鼠降低约14倍(n=3,P<0.01),对应结果见下图:
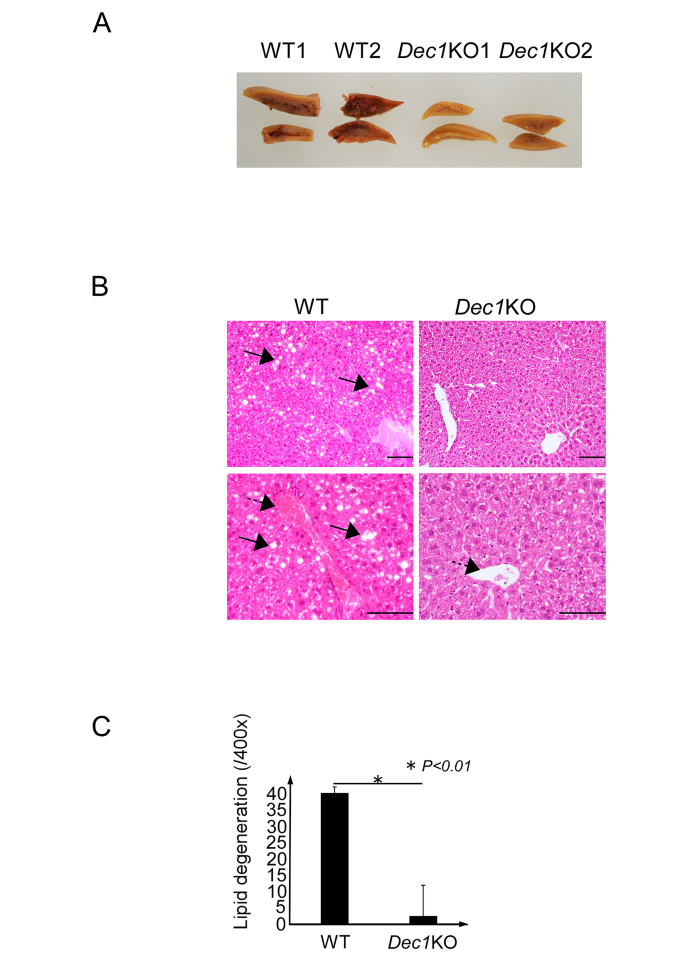
产品关联:文献未提及该环节具体实验产品,领域常规使用4%多聚甲醛固定液、石蜡包埋试剂、苏木精-伊红染色试剂盒、病理切片机、光学显微镜等。
3.4 代谢通路分子免疫组化检测
实验目的为探究Dec1缺失调控酒精诱导脂质蓄积的下游分子机制,检测AMPK和PPARs通路的表达变化。方法细节为采用免疫组化(IHC)检测肝脏组织中相关蛋白的表达,染色使用Ventana Discovery Auto-Stainer自动化平台进行,所用一抗包括:Cell Signaling Technology的磷酸化AMPK(pAMPK)兔单克隆抗体(货号2535,稀释比例1:300),圣克鲁斯生物技术(Santa Cruz Biotechnology)的总AMPK小鼠单克隆抗体(货号sc-74461,稀释比例1:50)、PPARα小鼠单克隆抗体(货号sc-398394,稀释比例1:50)、PPARγ小鼠单克隆抗体(货号sc-7273,稀释比例1:50),采用DAB法显色,显微镜下观察蛋白的表达水平与定位。结果解读显示,Dec1敲除小鼠肝脏中pAMPK的表达显著高于野生型小鼠,而总AMPK的表达在两组中无明显差异;同时Dec1敲除小鼠肝脏细胞核中PPARα的表达高于野生型,PPARγ的表达低于野生型,对应结果见下图:
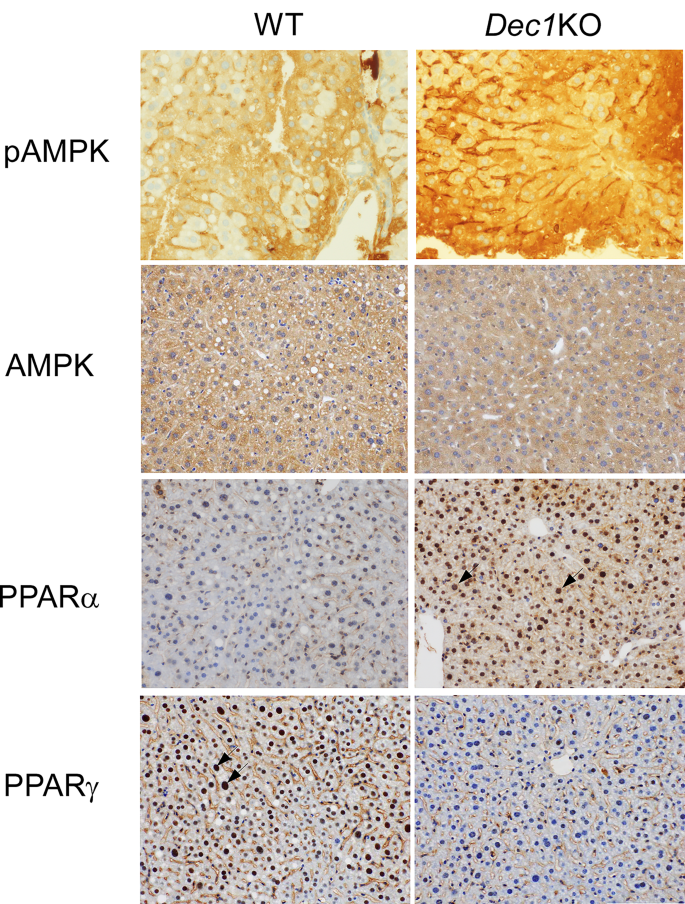
产品关联:实验所用关键产品:Cell Signaling Technology的磷酸化AMPK抗体(货号2535),Santa Cruz Biotechnology的总AMPK抗体(货号sc-74461)、PPARα抗体(货号sc-398394)、PPARγ抗体(货号sc-7273),Ventana Medical Systems的Discovery Auto-Stainer免疫组化平台及DAB-Map显色试剂盒(货号760-124)。
4. Biomarker 研究及发现成果
本研究涉及的功能性Biomarker为Dec1基因及其下游调控的磷酸化AMPK、PPARα、PPARγ,验证逻辑基于前期DEC1与上述通路的调控关系研究,通过动物表型验证后直接检测肝脏组织中上述分子的表达变化,明确其与酒精诱导肝脏损伤的关联。
上述Biomarker的检测样本为慢性酒精暴露3个月后的小鼠肝脏石蜡组织,验证方法为免疫组化染色,其中磷酸化AMPK在Dec1敲除小鼠肝脏中表达显著升高,总AMPK无明显组间差异;PPARα的核表达水平在敲除组中上调,PPARγ的表达在敲除组中下调。病理定量结果显示Dec1敲除小鼠的肝脏脂质变性位点较野生型降低14倍(n=3,P<0.01),但文献未提供该组Biomarker用于诊断或预后评估的ROC曲线、敏感性、特异性等相关数据。核心成果方面,本研究首次发现Dec1缺失可作为保护性因素,同时抑制慢性酒精诱导的肝脏脂质蓄积和昼夜节律紊乱,其分子机制与AMPK磷酸化激活、PPARα表达上调、PPARγ表达下调相关,所有组间差异均具有统计学显著性(P<0.01),实验样本量为酒精摄入实验每组9只、节律监测实验每组6只、病理和免疫组化分析每组3只。推测:Dec1可能是慢性酒精诱导肝脏损伤和节律紊乱的潜在干预靶点,后续可通过细胞实验进一步验证其调控机制,同时探究其在人类酒精性肝病患者中的表达差异,为临床转化提供依据。