1. 领域背景与文献引入
文献英文标题:Systematic analysis of gene expression in human brains before and after death;发表期刊:Genome Biology;影响因子:未公开;研究领域:神经科学(人类脑组织基因表达调控)
领域共识:微阵列技术已成为解析人类疾病、衰老及进化分子机制的核心工具,由于细胞培养和动物模型无法完全复现人类生理病理状态,死后人类组织尤其是脑组织成为这类研究的主要样本来源。此前研究已关注死后间隔、冷冻方法、储存时长等因素对基因表达的影响,发现这些因素的影响相对有限,但死亡本身是否会导致脑组织基因表达的系统性、功能性变化,以及这种变化是否会干扰脑区间固有表达差异的检测,这一核心问题尚未得到系统解答。部分小规模研究仅检测少量基因,提示死亡可能影响基因表达,但缺乏全基因组层面、多脑区的验证,也未排除手术样本的疾病干扰。因此,本研究旨在通过系统对比生前手术样本与死后样本的基因表达谱,明确死亡对人类脑组织基因表达的影响模式,为死后样本在神经科学研究中的应用提供严谨依据。
2. 文献综述解析
本文综述部分围绕“死后脑组织样本的基因表达可靠性”这一核心问题展开,以“现有研究的局限→待解决的核心问题→本研究的必要性”为逻辑主线。
现有研究已证实微阵列技术在神经科学研究中的应用价值,明确死后样本的质量控制因素(如死后间隔、冷冻方式)对基因表达的影响较小,但这些研究多聚焦于样本处理环节的干扰,未系统探讨死亡本身对基因表达的直接作用。此前仅有的小规模研究检测14个基因发现1个存在死亡诱导的表达变化,推测可能有大量基因受影响,但缺乏全基因组层面的验证,也未涉及多脑区的一致性分析。同时,现有研究未充分排除手术样本的疾病背景干扰,无法明确观察到的差异是源于死亡还是术前疾病。
本研究的创新点在于首次在全基因组层面、两个独立脑区中系统对比生前与死后样本的基因表达,通过多维度分析排除疾病干扰,明确死亡诱导的基因表达变化的功能特征及对脑区间差异检测的影响,填补了领域内关于死亡本身对脑组织基因表达影响的研究空白。
3. 研究思路总结与详细解析
本研究的整体框架为“提出假设→样本收集与检测→差异表达分析→功能验证→结论推导”,研究目标是明确死亡对人类脑组织基因表达的影响模式,验证死后样本用于脑区间基因表达差异研究的可靠性;核心科学问题包括死亡是否会引发系统性的基因表达变化、这种变化是否具有脑区一致性、是否会干扰脑区间固有表达差异的检测;技术路线为收集无神经疾病的死后脑组织样本与手术患者的正常邻近脑组织样本(覆盖额叶皮层、海马体两个脑区),采用Affymetrix微阵列检测基因表达谱,通过ANOVA、SAM等方法分析死亡诱导的差异表达基因,结合基因本体(GO)富集分析解析功能特征,同时通过多维度验证排除疾病干扰,最终评估死后样本的适用性。
3.1 样本收集与质量控制
实验目的:获取匹配的生前(手术)与死后脑组织样本,控制混杂因素(如神经疾病、RNA质量)以确保差异分析的可靠性。
方法细节:死后样本来自10名无神经疾病史、猝死的个体,采集额叶皮层和海马体组织;手术样本来自12名脑肿瘤或难治性癫痫患者,采集肿瘤/病灶旁的正常脑组织(经病理验证无异常),其中11名患者诊断为癫痫,1名无癫痫。所有样本均快速冷冻至-80℃,采用TRIZol试剂提取总RNA,经QIAGEN RNeasy试剂盒纯化,通过Agilent 2100生物分析仪检测RNA质量(28S/18S核糖体RNA比值),同时以微阵列上GAPDH基因的5"/3"信号比作为质量控制指标。
结果解读:所有样本的RNA质量均符合实验要求,28S/18S比值及GAPDH信号比均处于正常范围,确保后续基因表达检测的准确性。
产品关联:实验所用关键产品:TRIZol(GIBCO,货号未提及)、QIAGEN RNeasy试剂盒(货号未提及)、Agilent 2100 Bioanalyser、Affymetrix HG-U133plus2微阵列芯片。
3.2 基因表达谱检测与标准化分析
实验目的:获取两个脑区、两种样本类型的全基因组表达谱数据,并进行标准化处理以消除技术偏差。
方法细节:取1.2μg总RNA按照Affymetrix标准流程进行标记、杂交至HG-U133plus2芯片,完成染色、洗涤后扫描芯片获取原始信号数据;采用稳健多芯片平均(RMA)法进行数据标准化,转换为以2为底的对数表达值,同时计算检测P值筛选可检测到的探针集(P<0.065)。
结果解读:共检测到42427个可检测的探针集(占总探针集的77.69%),标准化后的数据符合正态分布,可用于后续差异表达分析。
产品关联:实验所用关键产品:Affymetrix HG-U133plus2微阵列芯片、Affymetrix GeneChip Operating Software(货号未提及)。
3.3 死亡诱导的基因表达差异分析
实验目的:明确死亡是否会导致脑组织基因表达的系统性变化,以及这种变化的脑区一致性。
方法细节:采用方差分析(ANOVA)和显著性分析微阵列(SAM)两种方法筛选差异表达探针集,ANOVA设置名义显著性阈值P<0.01(假发现率FDR=4.12%),SAM设置FDR=5%;同时通过线性回归和SAM分析排除癫痫对基因表达的影响,对比癫痫与非癫痫患者样本的差异表达相关性。
结果解读:ANOVA检测到5703个差异表达探针集(占可检测探针集的13.4%),其中96.7%与SAM结果重叠;这些差异中96%在额叶皮层和海马体两个脑区共享,提示死亡诱导的基因表达变化具有脑区一致性。进一步分析排除癫痫干扰:未发现癫痫对基因表达有显著影响的探针集,癫痫与非癫痫患者样本的差异表达相关性极高(Pearson相关系数R=0.948,n=2983,p<10^-15),证实观察到的差异主要源于死亡而非术前疾病。
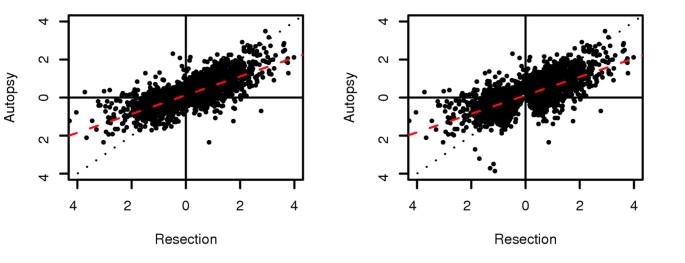
3.4 脑区间表达差异的死后保留性分析
实验目的:验证死后样本是否能保留生前脑区间的固有基因表达差异模式。
方法细节:采用ANOVA和SAM筛选脑区间的差异表达探针集,分别分析死后样本与手术样本中的脑区间差异,计算两种样本类型中脑区间表达差异的相关性,并通过线性回归分析差异幅度的变化。
结果解读:手术样本中检测到6568个脑区间差异探针集,死后样本中检测到的差异探针集数量减少,但两种样本类型中脑区间的表达差异模式高度相关(Pearson相关系数R=0.763,n=4471,p<10^-15);线性回归显示死后样本中脑区间差异的幅度平均为手术样本的50%,提示死后样本保留了脑区间的差异模式,但差异幅度显著降低。
3.5 死亡诱导差异的功能富集与变异贡献分析
实验目的:解析死亡诱导的差异表达基因的功能特征,以及这些差异对死后样本间表达变异的贡献。
方法细节:采用基因本体(GO)富集分析,对比差异表达基因与所有可检测基因的功能类别分布,通过二项式检验分析功能类别的上调/下调趋势;同时分析死亡诱导的差异基因在死后样本间的表达变异程度,对比其与其他基因的变异水平。
结果解读:死亡诱导的差异基因显著富集于蛋白质代谢、核酸代谢、细胞器组装等功能类别,其中泛素循环相关基因表达上调,蛋白质生物合成、rRNA加工相关基因表达下调;而细胞通讯、信号转导、发育等功能类别的基因表达在死后样本中保持稳定。此外,死亡诱导的差异基因在死后样本间的表达变异程度与其他基因无显著差异(Student"s t检验,p=0.916),提示这些差异不会显著影响死后样本间的表达变异分析。
产品关联:文献未提及具体功能富集分析工具,领域常规使用DAVID、STRING等生物信息学分析平台。
4. Biomarker研究及发现成果解析
本研究中涉及的Biomarker为死亡诱导的差异表达基因及脑区间固有表达差异特征,通过全基因组筛选、多脑区验证及疾病干扰排除,明确了其表达模式、功能特征及应用价值。
Biomarker定位:死亡诱导的差异表达基因(占可检测基因的13.4%),筛选逻辑为“微阵列全基因组检测→ANOVA与SAM双重差异分析→多脑区一致性验证→疾病干扰排除”;脑区间固有表达差异特征的验证逻辑为“手术样本差异检测→死后样本差异对比→相关性分析”。
研究过程详述:死亡诱导的差异基因来源于人类额叶皮层和海马体的死后与手术样本,验证方法包括ANOVA、SAM、GO富集分析、相关性分析,其特异性表现为96%的差异在两个脑区共享,敏感性为可检测出13.4%的基因存在死亡诱导的表达变化;脑区间表达差异的特异性表现为两种样本类型中的差异模式高度相关(R=0.763,n=4471,p<10^-15),敏感性为死后样本可检测到约50%幅度的差异。
核心成果提炼:死亡诱导的差异基因富集于基础代谢功能类别,泛素循环相关基因上调提示死后可能存在蛋白质降解增强,为死后样本的蛋白质组学研究提供了警示;脑区间的表达差异模式在死后样本中高度保留,但幅度降低50%,提示死后样本可用于脑区间差异研究,但需注意差异幅度的低估;死亡诱导的差异对死后样本间的表达变异无显著贡献,提示死后样本可用于群体水平的基因表达变异分析。统计学结果包括:死亡诱导的差异探针集数量为5703个(n=22,P<0.01,FDR=4.12%),脑区间差异的相关性R=0.763(n=4471,p<10^-15),死后样本差异幅度为手术样本的50%(回归斜率β=0.49)。