1. 领域背景与文献引入
文献英文标题:SRTsim: spatial pattern preserving simulations for spatially resolved transcriptomics;发表期刊:Genome Biology;影响因子:17.906(2022年);研究领域:空间转录组学计算工具开发
空间转录组学技术自2016年首个原位捕获技术(ST)问世以来,已发展出基于单分子荧光原位杂交(smFISH)、原位捕获、原位测序、显微切割四大类技术体系,涵盖10x Visium、MERFISH、seqFISH+等主流平台,为解析组织空间结构、细胞异质性、细胞间通讯等生物学问题提供了高分辨率研究手段,推动了肿瘤学、神经科学等多个领域的突破性进展。当前研究热点聚焦于开发针对空间转录组的专属计算分析方法,包括空间聚类、空间差异表达分析、细胞通讯推断等方向。然而,这类计算方法的开发与高度依赖标准化的模拟数据,现有模拟体系存在核心瓶颈:单细胞转录组模拟工具无法整合空间信息,无法保留空间转录组特有的基因空间表达模式;而现有空间转录组模拟多为特定研究中的自定义实现,存在记录不完整、可重复性差、模拟数据与真实数据契合度低的问题,且多数模拟基于待评估方法的假设,会引入偏倚导致方法性能被高估。因此,开发一个独立、可重复、能真实模拟空间转录组数据(同时保留基因表达特征与空间模式)的框架,成为空间转录组学计算方法发展的关键需求,本研究的核心初衷正是开发SRTsim工具以填补这一空白,为空间转录组计算方法的基准测试与实验设计提供可靠支撑。
2. 文献综述解析
作者首先系统梳理了空间转录组学的技术发展脉络,将现有技术按原理分为四大类并总结代表平台,随后针对模拟数据在计算方法开发中的核心价值,对现有模拟工具进行分类评述,明确了单细胞模拟工具与现有空间模拟的局限性。
现有研究已证实空间转录组技术对生物学研究的革新作用,同时明确模拟数据是计算方法开发、性能评估、实验设计的核心支撑。单细胞模拟工具(如ZINB-WaVE、scDesign2等)的优势在于能精准模拟基因表达的统计特征(如均值、方差、零比例),但核心局限性是无法整合空间信息,完全缺失空间转录组特有的基因空间表达模式;而现有空间转录组模拟多为零散的自定义实现,缺乏标准化流程,存在可重复性差、模拟数据与真实数据契合度低的问题,且部分模拟基于待评估方法的假设,会引入偏倚导致性能高估。本研究的创新价值在于首次开发了专门针对空间转录组的模拟工具SRTsim,突破了单细胞模拟工具的空间信息缺失瓶颈,既能保留基因表达的统计特征,又能精准还原基因的空间表达模式,同时支持参考型与无参考型模拟,适用于多种空间转录组平台,为空间转录组计算方法的公平、可靠评估提供了标准化框架。
3. 研究思路总结与详细解析
本研究的核心目标是开发一个可扩展、可重复、真实的空间转录组模拟工具SRTsim,核心科学问题是如何在模拟基因表达计数的同时,精准保留基因的空间表达模式;技术路线遵循“框架构建→性能验证→应用场景验证”的闭环逻辑:首先设计并实现SRTsim的模拟框架,支持参考型与无参考型模拟;随后通过与8种单细胞模拟工具对比,验证SRTsim生成数据与真实数据的一致性;最后在空间聚类、空间表达分析、细胞通讯推断三大典型应用场景中,验证SRTsim对计算方法基准测试的支撑能力。
3.1 SRTsim模拟框架的设计与实现
实验目的:构建一个灵活、通用的空间转录组模拟框架,能够同时保留基因表达的统计特征和空间表达模式,支持多种模拟场景与空间转录组平台。
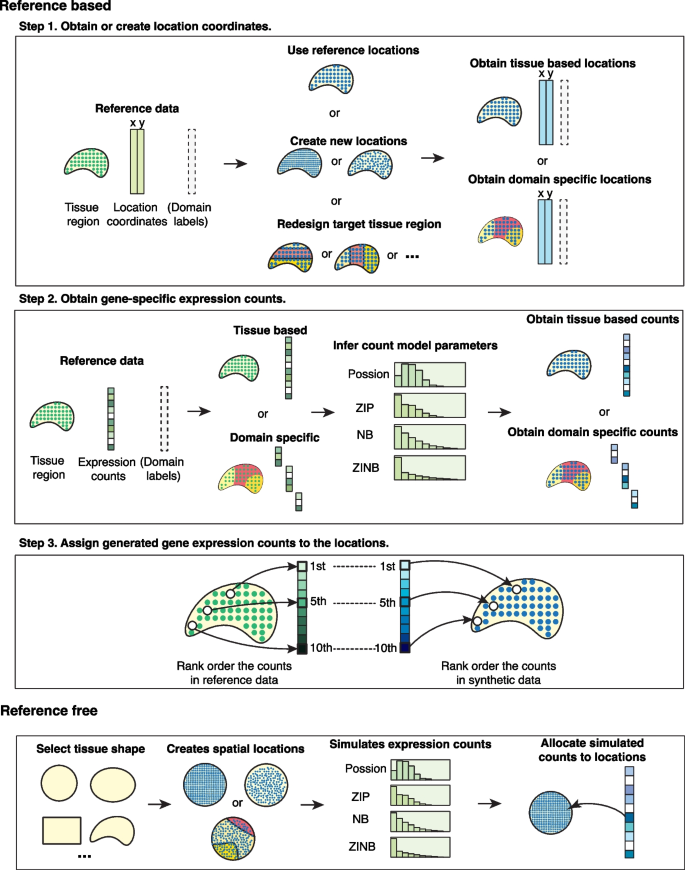
方法细节:SRTsim主要实现两种模拟模式:参考型模拟和无参考型模拟。参考型模拟分为三个核心步骤:第一步,获取或创建合成数据的空间位置坐标,可直接复用参考数据的位置,或根据用户需求生成新位置(通过凹壳算法拟合组织轮廓,再用网格或随机点过程生成指定数量的位置);第二步,针对每个基因,基于参考数据拟合四种计数模型(Poisson、零膨胀Poisson(ZIP)、负二项式(NB)、零膨胀负二项式(ZINB)),选择Akaike信息准则(AIC)最低的模型作为该基因的最优模型,基于模型参数生成表达计数;第三步,按照参考数据中基因的空间表达水平对位置排序,将模拟的表达计数按此顺序分配,确保保留空间模式。无参考型模拟允许用户自定义组织形状(可选预设形状或自定义轮廓)、位置数量、计数模型参数,生成具有指定空间模式的模拟数据。工具以R包形式实现,配套Shiny交互式应用,支持可视化模拟结果。
结果解读:SRTsim成功实现了两种模拟模式,输出结果为包含位置坐标矩阵、表达计数矩阵、模型参数的S4对象,结合用户指定的随机种子可确保模拟结果的完全可重复性;参考型模拟能精准还原参考数据的基因表达特征和空间模式,无参考型模拟可灵活生成符合实验设计需求的模拟数据。
产品关联:文献未提及具体实验产品,领域常规使用R语言及相关生物信息学工具包(如Splatter、Seurat、Giotto等)。
3.2 SRTsim模拟数据与真实数据的一致性验证
实验目的:验证SRTsim生成的模拟数据在基因表达统计特征和空间表达模式上与真实空间转录组数据的一致性,并与现有单细胞模拟工具进行对比。
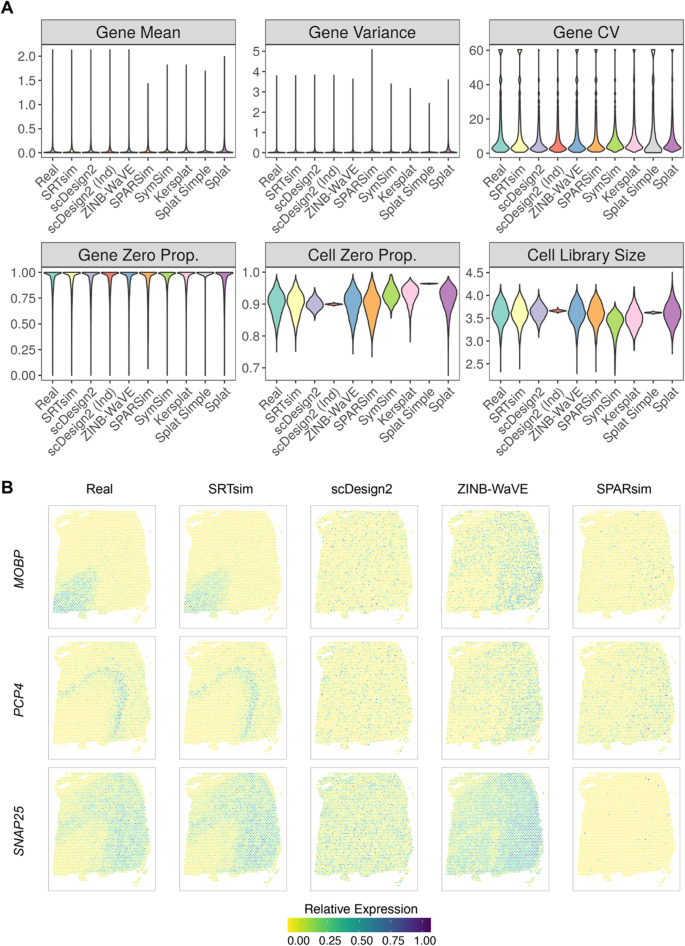
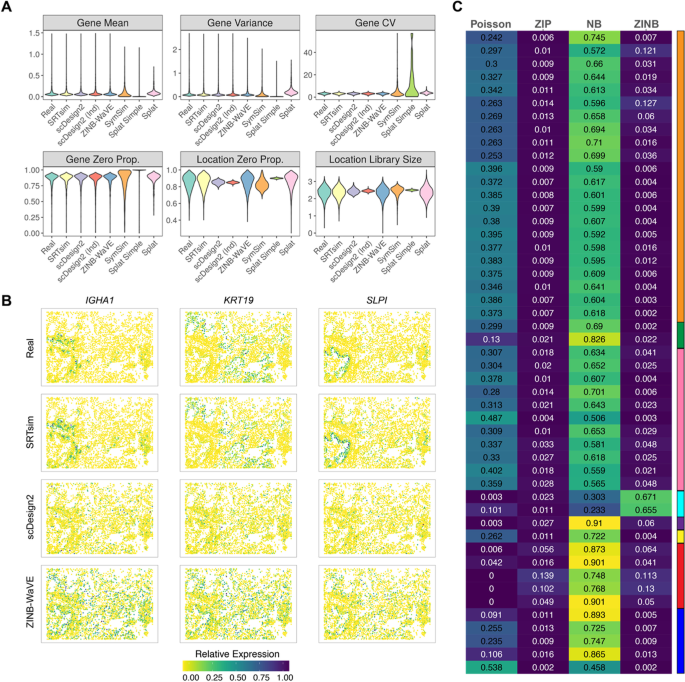
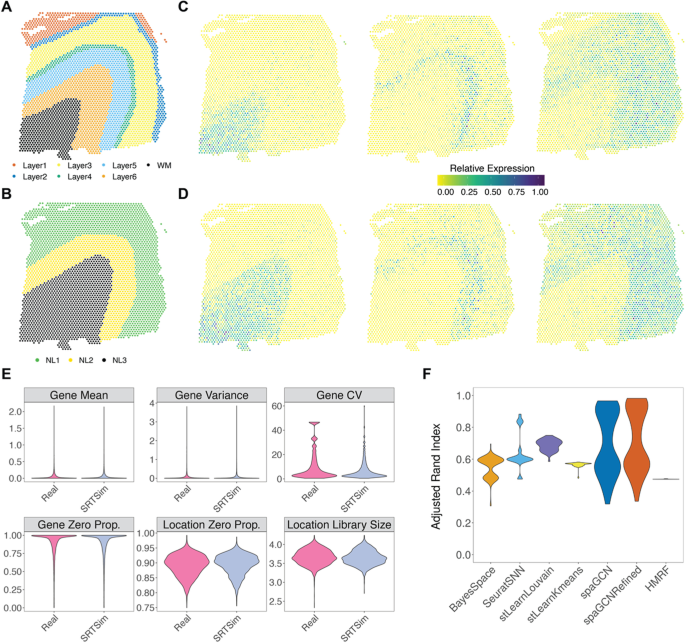
方法细节:收集来自8种空间转录组平台(10x Visium、MERFISH、seqFISH+等)的49个真实数据集作为参考,使用SRTsim和8种单细胞模拟工具(ZINB-WaVE、SPARSim、SymSim、scDesign2两种变体、Splat三种变体)分别进行参考型模拟;计算6个核心评估指标,包括4个基因水平指标(表达均值、方差、变异系数、零比例)和2个位置水平指标(零比例、文库大小),通过Kolmogorov–Smirnov检验对比模拟数据与真实数据的指标分布差异;通过可视化标记基因(如MOBP、PCP4、SNAP25)的空间模式、计算Moran"s I统计量(衡量空间自相关性)验证空间模式的保留效果;同时记录各工具的计算时间、内存占用及模拟成功率,评估计算效率和稳定性。
结果解读:仅SRTsim和ZINB-WaVE能同时保留基因水平和位置水平的所有统计特征,其中SRTsim的所有指标与真实数据无统计学差异(P>0.05,Kolmogorov–Smirnov检验),而其他工具的多数指标与真实数据存在显著差异;在空间模式保留方面,仅SRTsim能精准还原所有标记基因的空间表达模式,其生成数据的Moran"s I统计量与真实数据高度一致(相关系数>0.9);计算效率上,SRTsim比scDesign2、ZINB-WaVE快一个数量级,对于包含51649个位置的Slide-seqV2数据,SRTsim仅需约2小时,而scDesign2需64小时;稳定性上,SRTsim对所有49个数据集的模拟成功率为100%,而SPARSim、Kersplat等工具存在模拟失败的情况。
3.3 SRTsim在空间聚类方法基准测试中的应用
实验目的:验证SRTsim生成的模拟数据可用于空间聚类方法的性能评估,分析不同实验参数和组织结构对聚类性能的影响。
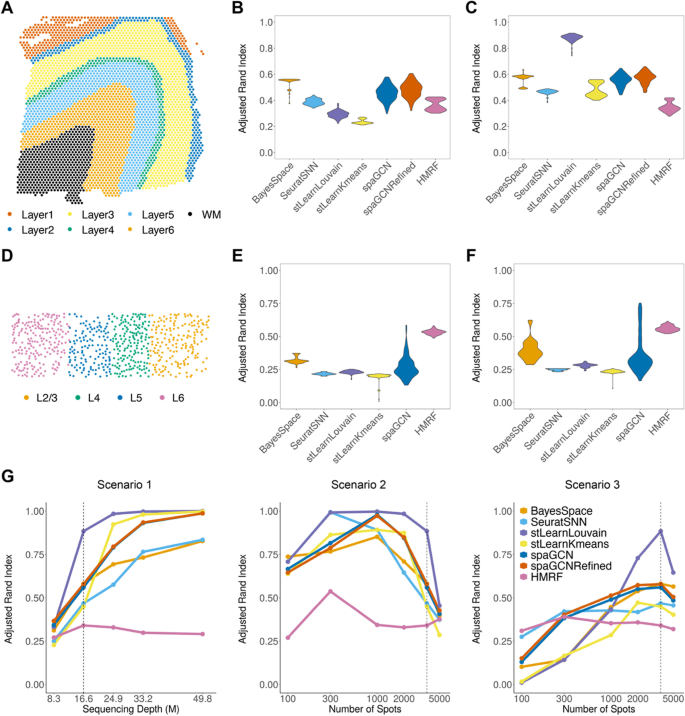
方法细节:选择两个代表性数据集作为参考:非单细胞分辨率的10x Visium人类背外侧前额叶皮层(DLPFC)数据(包含7个组织区域)和单细胞分辨率的STARmap小鼠视觉皮层数据(包含4个皮层层);分别生成组织水平(全组织模拟)和区域水平(分区域模拟后合并)的模拟数据;评估6种主流空间聚类方法(BayesSpace、stLearn Kmeans/Louvain、SpaGCN、HMRF、Seurat SNN)的性能,采用调整兰德指数(ARI)衡量聚类结果与真实区域的一致性;同时设计三种实验参数场景:(I)改变总测序深度、固定位置数量;(II)改变位置数量、固定总测序深度;(III)固定平均测序深度、同时改变位置数量和总测序深度,分析参数对聚类性能的影响;此外,通过合并DLPFC的原始7个区域为3个新区域,分析区域数量和大小对聚类性能的影响。
结果解读:不同聚类方法在不同数据集和模拟模式下的性能存在差异:在DLPFC组织水平模拟数据中,BayesSpace的ARI均值最高,达0.54(n=100,P<0.05);在STARmap组织水平模拟数据中,HMRF的ARI均值最高,达0.53(n=100,P<0.05);区域水平模拟数据的聚类性能普遍优于组织水平,如stLearn Louvain在DLPFC区域水平模拟中的ARI均值达0.88(n=100,P<0.05);参数分析显示,聚类性能随测序深度和位置数量增加先提升后饱和或下降,例如当DLPFC数据的测序深度达16.6M reads、位置数量达1000时,聚类性能接近最优;区域数量和大小显著影响聚类性能,合并区域后的模拟数据中,stLearn Louvain的ARI均值达0.70(n=10,P<0.05),而SpaGCN的性能存在批次差异。
3.4 SRTsim在空间表达分析方法基准测试中的应用
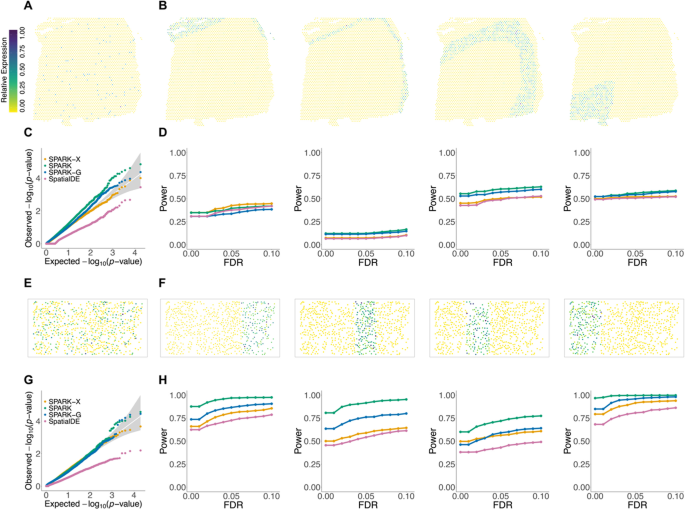
实验目的:验证SRTsim可用于空间表达(SE)分析方法的性能评估,分析信号强度、区域大小对空间表达基因检测性能的影响。
方法细节:基于DLPFC和STARmap数据生成两种模拟场景:零假设场景(1000个无空间模式的基因)和备择假设场景(900个无空间模式基因+100个具有区域特异性空间模式的基因),针对备择假设场景设置不同的信号强度(如5倍、10倍表达差异);评估4种主流SE分析方法(SPARK、SPARK-G、SPARK-X、SpatialDE)的性能,零假设场景下通过Q-Q图验证P值校准度,备择假设场景下通过FDR-功效曲线衡量检测性能;同时分析不同区域大小(如DLPFC的薄层2、4与厚层)对检测性能的影响。
结果解读:零假设场景下,SPARK、SPARK-G、SPARK-X的P值校准良好(Q-Q图接近对角线),而SpatialDE的P值过于保守;备择假设场景下,SPARK和SPARK-G在多数空间模式下的检测功效更高,例如在DLPFC的白质区域模式中,FDR=0.05时功效达0.85以上,而SPARK-X在薄层1的模式中表现更优;检测性能依赖信号强度和区域大小,信号越强、区域越大,检测功效越高,例如DLPFC的薄层2中,信号强度为10倍时的功效是5倍时的1.5倍以上。
3.5 SRTsim在细胞通讯推断方法基准测试中的应用
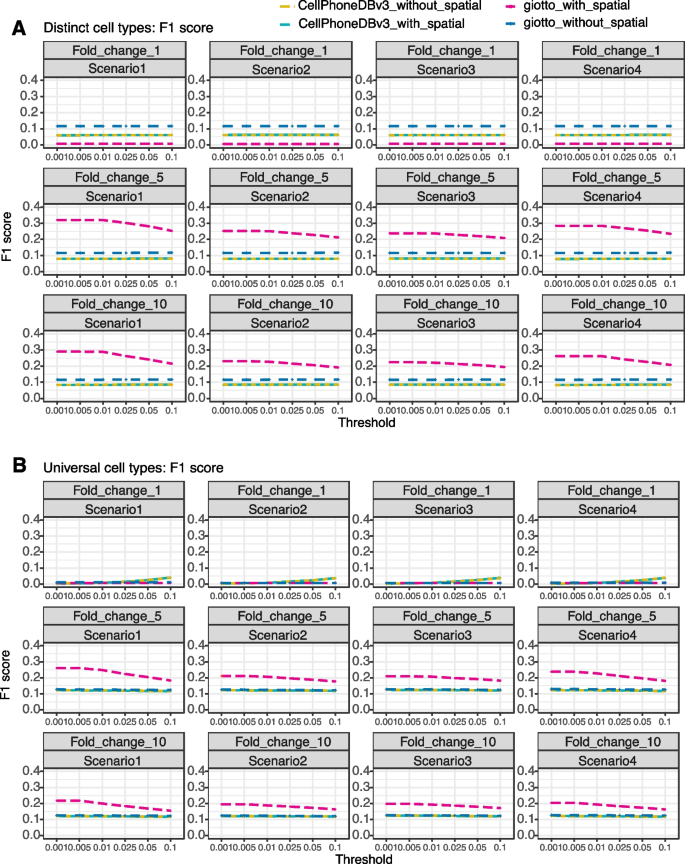
实验目的:验证SRTsim可用于单细胞分辨率空间转录组的细胞通讯推断方法性能评估,比较不同方法的检测能力。
方法细节:基于STARmap数据生成单细胞分辨率的模拟数据,设置同质基因表达(所有细胞的基因表达来自同一分布)和异质基因表达(细胞类型特异性表达分布)两种场景,同时设置无通讯、中度通讯、高度通讯三种信号强度;评估4种方法的性能:Giotto带/不带空间信息、CellphoneDBv3带/不带空间信息,采用F1值(精确率与召回率的调和均值)衡量综合性能,同时评估精确率、召回率、特异性。
结果解读:带空间信息的Giotto综合性能最优,在异质表达的中度通讯场景中,F1值均值达0.204(n=10,P<0.05);不带空间信息的Giotto召回率最高,而CellphoneDBv3的特异性最高;空间信息的引入显著提升了Giotto的性能,而对CellphoneDBv3的影响较小,说明不同方法对空间信息的利用效率存在差异。
4. Biomarker研究及发现成果解析
本研究虽未直接发现新的Biomarker,但通过SRTsim工具的开发与应用,为空间特异性表达Biomarker的研究提供了标准化的评估平台,支持空间表达Biomarker检测方法的性能验证,间接助力空间Biomarker的筛选与验证。
Biomarker定位:涉及的Biomarker类型为空间特异性表达基因(如人类DLPFC中的MOBP、PCP4、SNAP25,小鼠视觉皮层中的层特异性基因),筛选/验证逻辑为:基于真实空间转录组数据构建参考→用SRTsim生成保留空间模式的模拟数据→采用SPARK等空间表达分析方法检测空间特异性基因→验证模拟数据中检测到的基因空间模式与真实数据一致。
研究过程详述:Biomarker来源为真实空间转录组数据集(如人类DLPFC、小鼠视觉皮层),验证方法为利用SRTsim生成模拟数据,通过空间表达分析方法检测空间特异性基因,特异性与敏感性数据显示:在DLPFC模拟数据中,SPARK在FDR=0.05时检测空间特异性基因的功效可达0.8以上,模拟数据的Moran"s I统计量与真实数据的相关系数>0.9,表明模拟数据能真实反映基因的空间特异性;在STARmap模拟数据中,SPARK检测层特异性基因的功效在0.6以上(FDR=0.05)。
核心成果提炼:SRTsim为空间特异性表达Biomarker的研究提供了可靠的模拟评估平台,能够支持检测方法的性能验证,帮助筛选出真实的空间特异性基因,这类基因可作为组织区域划分、细胞类型鉴定的Biomarker;创新性在于首次实现了保留空间模式的空间转录组模拟,解决了现有模拟工具无法支撑空间Biomarker评估的瓶颈;研究未提供风险比(HR)数据,但通过功效、ARI、Moran"s I等指标验证了模拟数据的可靠性,为空间Biomarker的标准化研究奠定了基础。