Abstract
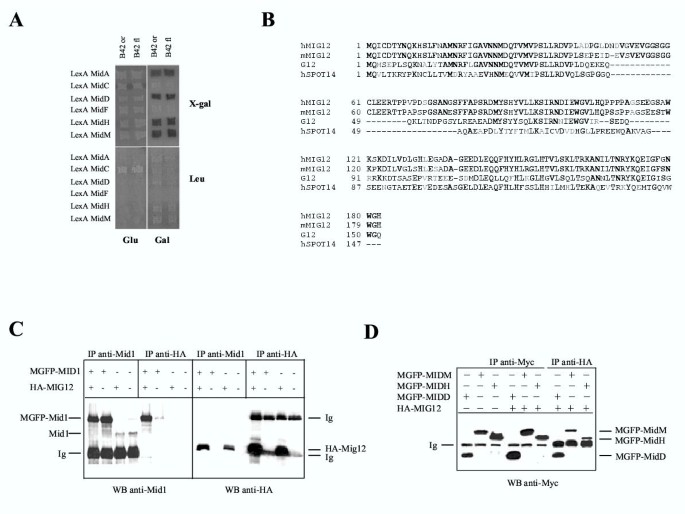
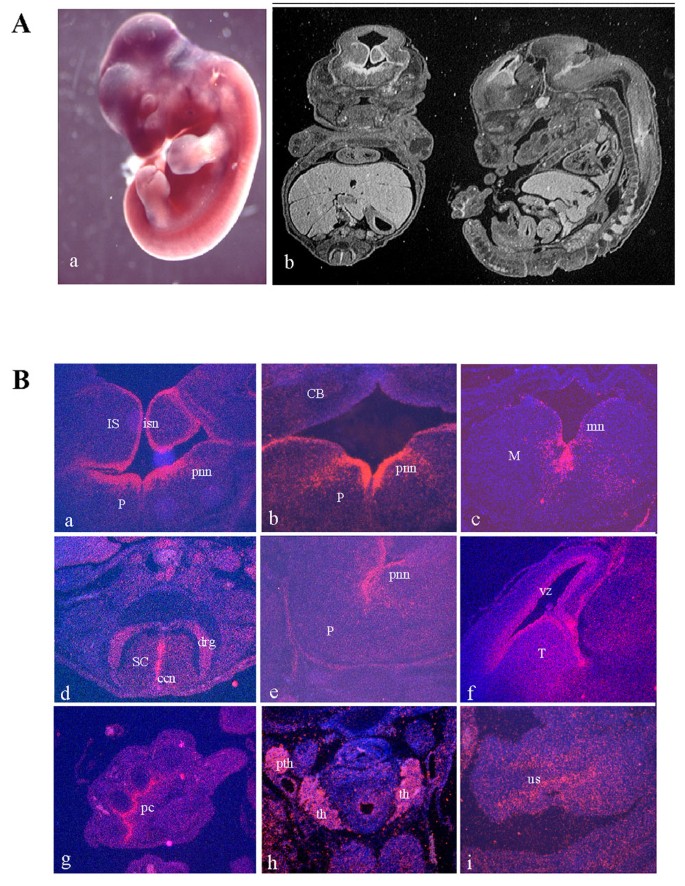
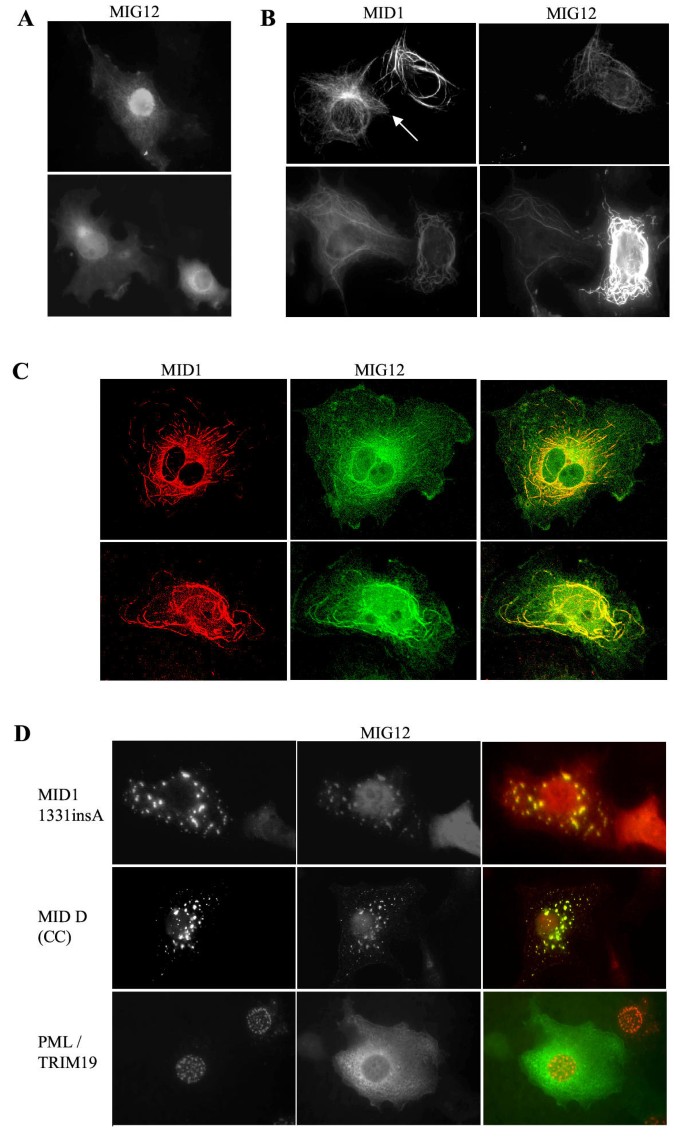
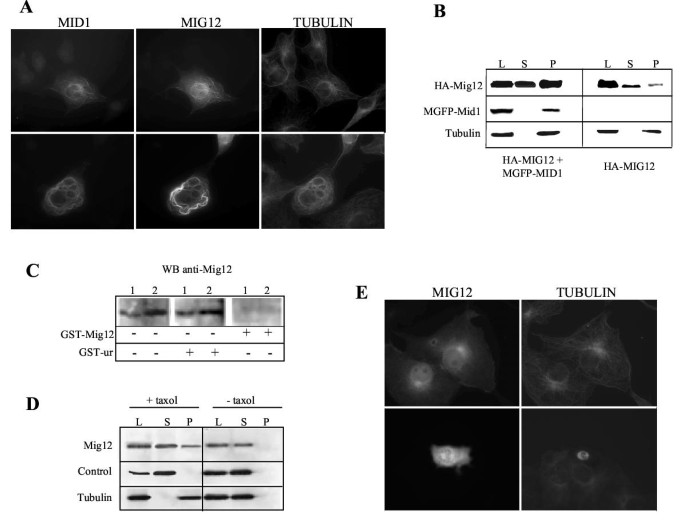
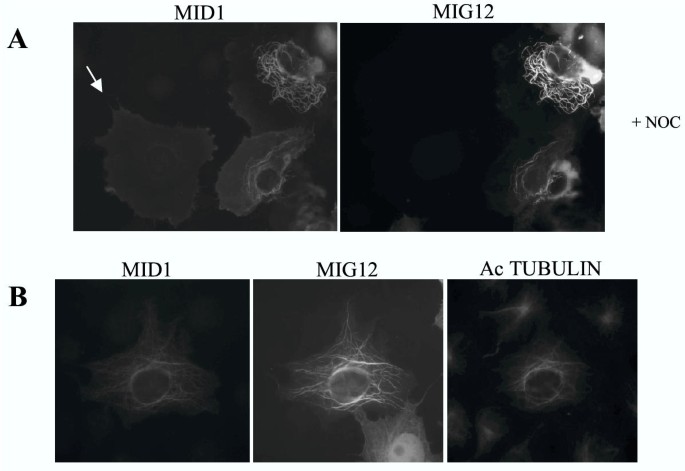
BACKGROUND: Opitz G/BBB syndrome is a genetic disorder characterized by developmental midline abnormalities, such as hypertelorism, cleft palate, and hypospadias. The gene responsible for the X-linked form of this disease, MID1, encodes a TRIM/RBCC protein that is anchored to the microtubules. The association of Mid1 with the cytoskeleton is regulated by dynamic phosphorylation, through the interaction with the alpha4 subunit of phosphatase 2A (PP2A). Mid1 acts as an E3 ubiquitin ligase, regulating PP2A degradation on microtubules. RESULTS: In spite of these findings, the biological role exerted by the Opitz syndrome gene product is still unclear and the presence of other potential interacting moieties in the Mid1 structure prompted us to search for additional cellular partners. Through a yeast two-hybrid screening approach, we identified a novel gene, MIG12, whose protein product interacts with Mid1. We confirmed by immunoprecipitation that this interaction occurs in vivo and that it is mediated by the Mid1 coiled-coil domain. We found that Mig12 is mainly expressed in the neuroepithelial midline, urogenital apparatus, and digits during embryonic development. Transiently expressed Mig12 is found diffusely in both nucleus and cytoplasm, although it is enriched in the microtubule-organizing center region. Consistently with this, endogenous Mig12 protein is partially detected in the polymerized tubulin fraction after microtubule stabilization. When co-transfected with Mid1, Mig12 is massively recruited to thick filamentous structures composed of tubulin. These microtubule bundles are resistant to high doses of depolymerizing agents and are composed of acetylated tubulin, thus representing stabilized microtubule arrays. CONCLUSIONS: Our findings suggest that Mig12 co-operates with Mid1 to stabilize microtubules. Mid1-Mig12 complexes might be implicated in cellular processes that require microtubule stabilization, such as cell division and migration. Impairment in Mig12/Mid1-mediated microtubule dynamic regulation, during the development of embryonic midline, may cause the pathological signs observed in Opitz syndrome patients.