1. 领域背景与文献引入
文献英文标题:Use of high-density tiling microarrays to identify mutations globally and elucidate mechanisms of drug resistance in Plasmodium falciparum;发表期刊:Genome Biology;影响因子:未公开;研究领域:疟疾分子生物学、抗疟药耐药机制研究。
恶性疟原虫是导致人类重症疟疾的主要病原体,其高度的遗传变异性和性重组能力推动了抗疟药耐药性的快速出现与传播,导致氯喹、甲氟喹等多种传统抗疟药疗效下降。领域发展关键节点包括2002年恶性疟原虫全基因组测序完成,为耐药机制研究提供了基因组基础;传统耐药基因鉴定方法如遗传杂交、候选基因法存在耗时久、成本高、无法覆盖全基因组的局限,尤其是对于作用机制不明的新型抗疟药,难以快速定位耐药相关遗传变异。当前研究热点集中在全基因组水平解析耐药机制、发现新型耐药生物标志物,而未解决的核心问题包括缺乏高效的全基因组遗传变异检测手段,以及部分新型抗疟药的耐药机制尚未明确。
针对这一空白,本研究开发了基于高密度平铺微阵列的全基因组变异检测方法,旨在快速鉴定恶性疟原虫的单核苷酸多态性(SNP)和拷贝数变异(CNV),并解析抗疟药的耐药机制,为疟疾治疗和耐药监测提供技术支撑。
2. 文献综述解析
作者按传统遗传方法、候选基因法、新型基因组技术的时间线和技术维度,对恶性疟原虫耐药基因鉴定方法进行分类评述。
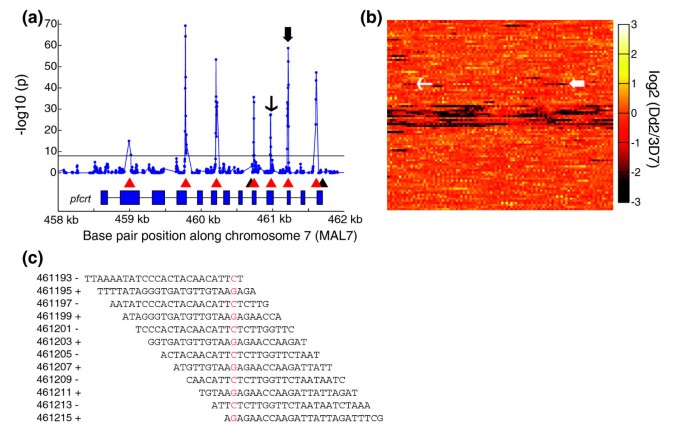
传统遗传方法如遗传杂交,曾成功鉴定氯喹耐药的关键基因pfcrt,但需依赖非人灵长类动物模型,成本高、周期长;啮齿类疟疾模型虽成本较低,但耐药机制无法完全外推至人类疟疾。候选基因法基于其他物种的耐药机制,成功发现pfmdr1基因扩增与甲氟喹耐药相关、pfdhfr-ts基因突变与抗叶酸类药物耐药相关,但该方法仅适用于作用机制已知的药物,对新型抗疟药的耐药基因鉴定存在局限性。新型基因组技术如全基因组重测序可检测SNP,但无法有效检测CNV,而CNV是恶性疟原虫应对药物压力的重要遗传变异类型。
现有方法存在全基因组覆盖不足、无法同时检测SNP和CNV的缺陷,本研究开发的高密度平铺微阵列方法可在单次杂交实验中同时检测SNP和CNV,且能精确定位CNV的断点,弥补了传统方法的不足,为全基因组水平解析耐药机制提供了高效工具。
3. 研究思路总结与详细解析
本研究的整体框架为:以开发全基因组遗传变异检测技术为核心,先完成高密度平铺微阵列的设计与构建,再通过已知变异株验证方法的准确性,随后将技术应用于工程虫系的意外突变检测和福斯霉素耐药机制解析,最终开发配套分析软件形成完整技术体系。研究目标是建立高效的恶性疟原虫全基因组变异检测方法,解析福斯霉素的耐药机制;核心科学问题是如何利用微阵列技术同时检测SNP和CNV,以及福斯霉素耐药的分子基础;技术路线遵循“方法构建→验证→应用→工具开发”的闭环逻辑。
3.1 高密度平铺微阵列设计与构建
实验目的是构建覆盖恶性疟原虫3D7株全基因组的高密度平铺微阵列,为全基因组变异检测提供平台。方法细节为基于3D7株参考基因组,设计包含480万条25mer探针的定制微阵列,特征尺寸为5μm,覆盖约90%的编码区和60%的非编码区,受限于恶性疟原虫基因组高AT含量,部分区域无法设计有效探针。结果解读显示,微阵列覆盖了基因组大部分功能区域,为后续变异检测奠定了基础。文献未提及具体实验产品,领域常规使用Affymetrix等品牌的定制微阵列及配套杂交、扫描设备。
3.2 微阵列方法学验证(CNV检测)
实验目的是验证微阵列检测基因扩增和缺失等拷贝数变异的准确性。方法细节为选取3D7、Dd2、HB3三个实验室株系,提取基因组DNA进行微阵列杂交,采用匹配积分分布(MOID)算法检测基因缺失,通过滑动窗口z检验检测基因扩增,以已知的pfmdr1和pfgch1基因为验证靶点。结果解读显示,MOID算法成功检测到Dd2株中6个端粒黏附基因的缺失,与已有报道一致;滑动窗口z检验成功定位Dd2株中pfmdr1基因的扩增区域,log₂比值约为0.7,提示存在3个拷贝,与定量PCR结果一致;同时精确定位了pfmdr1和pfgch1基因扩增的断点,误差在数百碱基以内。
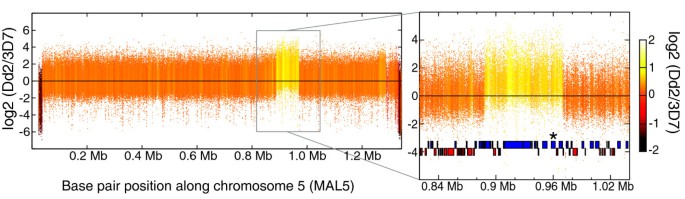
文献未提及具体实验产品,领域常规使用定量PCR试剂盒、MOID算法配套分析软件等。
3.3 微阵列方法学验证(SNP检测)
实验目的是验证微阵列检测单核苷酸多态性的准确性。方法细节为以Broad Institute测序的Dd2和HB3株SNP数据为金标准,采用滑动窗口z检验和F检验分析微阵列数据,筛选P值<1×10⁻⁸的探针作为SNP候选位点。结果解读显示,在Dd2株中检测到91.1%的已知SNP(1582个真阳性,155个假阴性),假发现率为10.5%;在HB3株中检测到85.0%的已知SNP(2842个真阳性,502个假阴性),假发现率为16.7%;进一步测序验证显示,约50%的“假阳性”实际为未被测序项目检测到的多态性,实际假发现率约为6%。
文献未提及具体实验产品,领域常规使用Sanger测序试剂盒、PCR扩增试剂等。
3.4 工程虫系的突变检测
实验目的是验证微阵列在检测工程虫系意外突变中的应用价值。方法细节为对3D7^{attB}工程虫系(插入attB重组位点和人dhfr筛选标记)进行微阵列杂交,分析其与亲本3D7株的遗传变异。结果解读显示,检测到6号染色体端粒区域的缺失(1.38-1.41Mb),以及组蛋白2B基因(PF11_0062)中的两个点突变,导致A100F的非同义突变,提示工程虫系在转染和筛选过程中可能产生意外突变,全基因组检测有助于排除这些突变对表型的干扰。
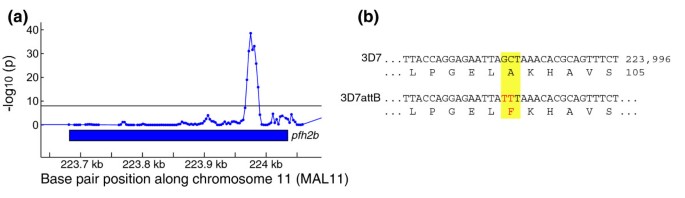
文献未提及具体实验产品,领域常规使用转染试剂、筛选药物等。
3.5 福斯霉素耐药机制解析
实验目的是解析恶性疟原虫对福斯霉素的耐药机制。方法细节为通过体外逐步增加药物浓度的方法,从Dd2株中筛选获得两株福斯霉素耐药克隆FOS-R^{Dd2-CL1}和FOS-R^{Dd2-CL2};采用[³H]次黄嘌呤掺入法检测耐药性;通过微阵列杂交分析耐药株与亲本株的遗传变异;采用定量PCR和定量RT-PCR验证基因拷贝数和转录水平;对pfdxr和pfdxs基因进行测序。结果解读显示,耐药株的福斯霉素半数抑制浓度(IC₅₀)分别为2219±136nmol/L和2623±78nmol/L,是亲本株(307±49nmol/L)的7-8倍(n=5-7,P<0.001);微阵列检测到耐药株14号染色体上约100kb的区域扩增,包含23个基因,其中pfdxr基因(福斯霉素的靶点,编码1-脱氧-D-木酮糖5-磷酸还原异构酶)位于该区域;定量PCR显示FOS-R^{Dd2-CL1}株pfdxr基因拷贝数增加2.7倍,定量RT-PCR显示转录水平增加3.8倍(n=2,P<0.001);测序未发现pfdxr和pfdxs基因的突变,提示pfdxr基因扩增是福斯霉素耐药的分子机制。
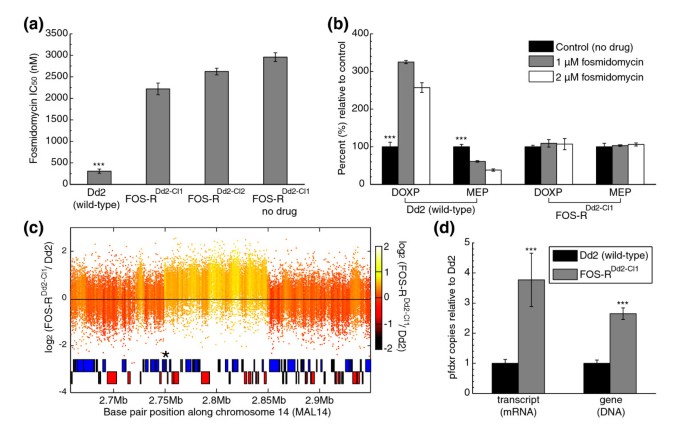
文献未提及具体实验产品,领域常规使用[³H]次黄嘌呤、定量PCR试剂盒、测序试剂等。
3.6 配套分析软件开发
实验目的是开发用于微阵列数据解析的可视化工具,降低技术使用门槛。方法细节为基于MATLAB开发图形用户界面(GUI)软件,实现SNP和CNV的可视化、SNP预测等功能。结果解读显示,软件可高效解析微阵列数据,为研究者提供直观的遗传变异可视化结果,且可免费获取。文献未提及具体实验产品,领域常规使用MATLAB软件及相关生物信息学工具包。
4. Biomarker研究及发现成果解析
Biomarker定位为pfdxr基因拷贝数变异,属于耐药性生物标志物;筛选/验证逻辑为“体外耐药株筛选→全基因组微阵列检测CNV→定量PCR验证基因拷贝数→定量RT-PCR验证转录水平→测序排除基因突变”的完整链条。
研究过程详述:该Biomarker来源为体外诱导的福斯霉素耐药恶性疟原虫株的基因组DNA;验证方法包括高密度平铺微阵列杂交、定量PCR、定量RT-PCR;特异性与敏感性数据显示,微阵列检测到的pfdxr基因扩增区域在两株耐药株中均存在,定量PCR显示基因拷贝数增加2.7倍(n=2,P<0.001),定量RT-PCR显示转录水平增加3.8倍(n=2,P<0.001),原文未提供ROC曲线数据。
核心成果提炼:该Biomarker的功能关联为pfdxr基因扩增通过增加靶点基因的表达量,克服福斯霉素对异戊二烯合成通路的抑制,从而介导耐药性;创新性为首次在恶性疟原虫中发现药物靶点基因扩增介导的福斯霉素耐药,填补了福斯霉素耐药机制研究的空白;统计学结果显示基因拷贝数和转录水平的差异均具有显著性(P<0.001,n=2),为福斯霉素的临床应用和耐药监测提供了重要依据。