Background
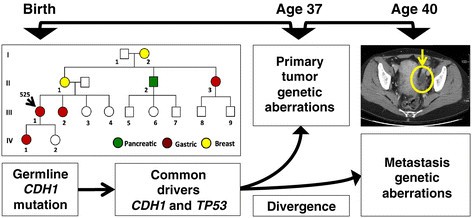
Gastric cancer is the second-leading cause of global cancer deaths, with metastatic disease representing the primary cause of mortality. To identify candidate drivers involved in oncogenesis and tumor evolution, we conduct an extensive genome sequencing analysis of metastatic progression in a diffuse gastric cancer. This involves a comparison between a primary tumor from a hereditary diffuse gastric cancer syndrome proband and its recurrence as an ovarian metastasis.
Conclusions
We document the metastatic differentiation and genetic heterogeneity of diffuse gastric cancer and reveal the potential metastatic role of TGFBR2 loss-of-function. In support of this study, we apply a murine primary organoid culture method capable of recapitulating in vivo metastatic gastric cancer. Overall, we describe an integrated approach to identify and functionally validate putative cancer drivers involved in metastasis.
Results
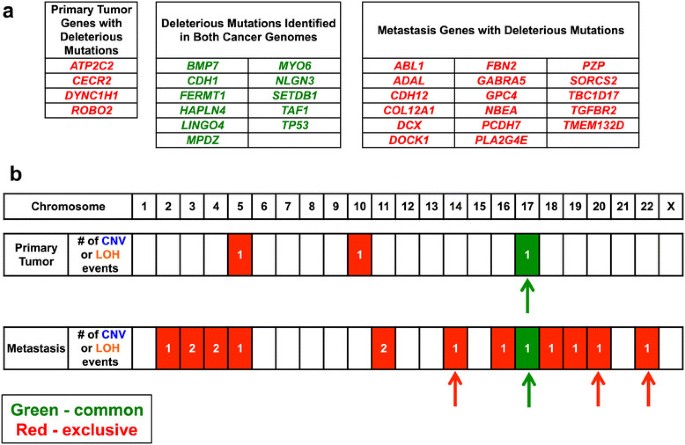
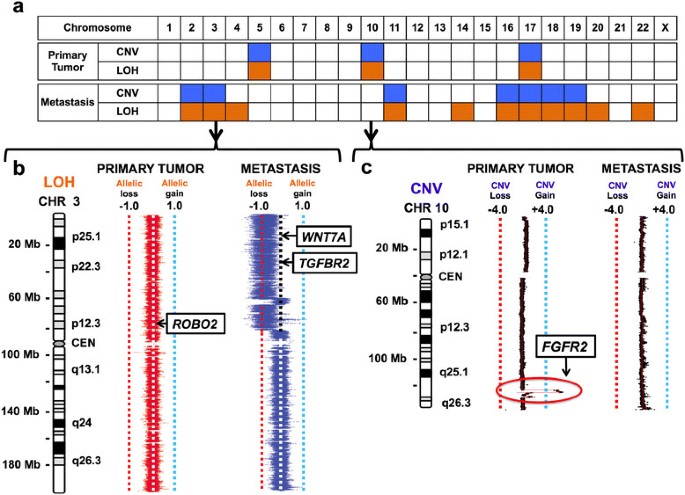
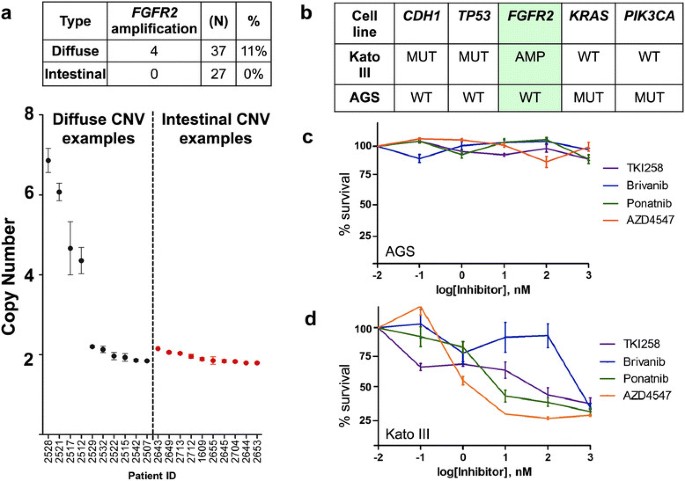
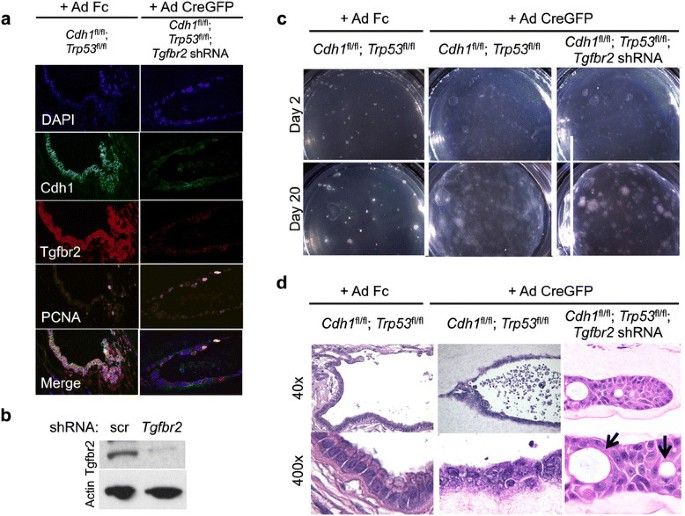
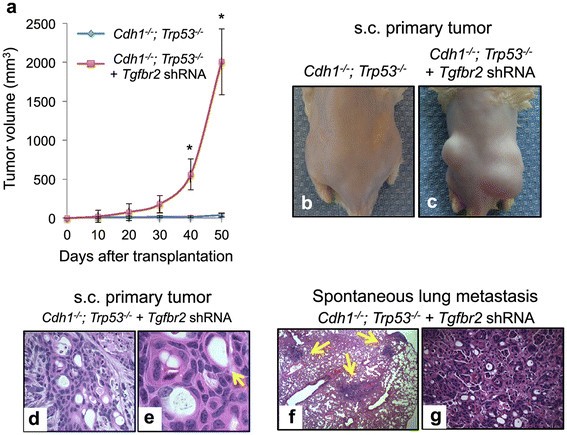
Both the primary tumor and ovarian metastasis have common biallelic loss-of-function of both the CDH1 and TP53 tumor suppressors, indicating a common genetic origin. While the primary tumor exhibits amplification of the Fibroblast growth factor receptor 2 (FGFR2) gene, the metastasis notably lacks FGFR2 amplification but rather possesses unique biallelic alterations of Transforming growth factor-beta receptor 2 (TGFBR2), indicating the divergent in vivo evolution of a TGFBR2-mutant metastatic clonal population in this patient. As TGFBR2 mutations have not previously been functionally validated in gastric cancer, we modeled the metastatic potential of TGFBR2 loss in a murine three-dimensional primary gastric organoid culture. The Tgfbr2 shRNA knockdown within Cdh1-/-; Tp53-/- organoids generates invasion in vitro and robust metastatic tumorigenicity in vivo, confirming Tgfbr2 metastasis suppressor activity. Conclusions: We document the metastatic differentiation and genetic heterogeneity of diffuse gastric cancer and reveal the potential metastatic role of TGFBR2 loss-of-function. In support of this study, we apply a murine primary organoid culture method capable of recapitulating in vivo metastatic gastric cancer. Overall, we describe an integrated approach to identify and functionally validate putative cancer drivers involved in metastasis.