1. 领域背景与文献引入
文献英文标题:Molecular signatures of chronic myeloid leukemia stem cells;发表期刊:Biomark Res;影响因子:未公开;研究领域:慢性髓性白血病干细胞分子标志物研究
慢性髓性白血病(CML)是由费城染色体(Ph⁺)编码的BCR-ABL融合基因驱动的 myeloproliferative disorder,其病程分为慢性期、加速期和急变期。2001年,伊马替尼(imatinib)作为首个BCR-ABL酪氨酸激酶抑制剂(TKI)获批用于CML治疗,显著提高了患者的5年完全细胞遗传学缓解率(达87%),但无法根除白血病干细胞(LSCs)——这是患者停药后复发的核心原因。现有研究表明,LSCs对TKIs耐药的关键在于其存活不依赖BCR-ABL激酶活性,且LSCs与分化的白血病细胞具有不同的分子调控机制。当前领域的核心问题是:对LSCs的分子特征认识零散,缺乏能监测LSCs活性、靶向LSCs的特异性标志物。因此,系统识别CML LSCs的新型分子标志物,解析其调控机制,成为治愈CML的关键突破口。本文正是针对这一需求,整合多维度基因(代谢、发育、激酶、黏附、转录),系统刻画CML LSCs的分子特征。
2. 文献综述解析
作者围绕“CML LSCs的生物学特性→TKIs耐药机制→现有标志物研究的局限性”展开综述,核心逻辑是“功能验证→机制探讨→需求导出”。
现有研究的关键结论包括:①CML由BCR-ABL阳性造血干细胞克隆扩增引起,LSCs具有自我更新、分化和体内成瘤能力;②LSCs对TKIs耐药的原因并非BCR-ABL激酶域突变,而是其存活依赖非激酶活性通路;③技术方法上,BCR-ABL逆转录病毒骨髓转导/移植小鼠模型是研究LSCs的核心工具。现有研究的优势是建立了LSCs的体内功能验证体系,局限性是对LSCs的分子调控网络认识零散,缺乏涵盖多功能类型的LSCs分子标志物谱——多数研究仅聚焦单一通路(如Wnt/β-catenin),未形成系统的标志物集合。
本文的创新价值在于:首次整合细胞代谢调控因子、HSC发育相关基因、激酶信号分子、细胞黏附分子、转录因子五大类基因,系统构建CML LSCs的分子标志物谱,弥补了现有研究对LSCs分子特征“碎片化”认识的不足,为LSCs的监测和靶向治疗提供了全面的候选靶点。
3. 研究思路总结与详细解析
整体框架
研究目标:系统识别CML LSCs的新型分子标志物,解析其调控机制;核心科学问题:LSCs对TKIs耐药的分子机制及潜在标志物;技术路线:“CML小鼠模型构建→LSCs分离与功能验证→多维度基因筛选→功能验证→临床样本验证”,即通过小鼠模型模拟CML发病,筛选LSCs差异基因,验证其对LSCs功能的调控,最终用临床样本确认其相关性。
3.1 CML LSCs的生物学特性验证
实验目的:明确BCR-ABL阳性造血干细胞作为LSCs的功能及对TKIs的耐药性。
方法细节:采用BCR-ABL逆转录病毒骨髓转导/移植小鼠模型,分离Lin⁻c-Kit⁺Sca-1⁺细胞(LSCs候选群),通过体内成瘤实验验证其诱导CML的能力;同时用伊马替尼处理,检测LSCs的存活情况。
结果解读:Lin⁻c-Kit⁺Sca-1⁺细胞能在受体小鼠中诱导典型CML表型(粒细胞增多、脾肿大),且伊马替尼处理后该细胞群仍存活(存活比例高于分化的白血病细胞),证明其作为LSCs的功能及对TKIs的耐药性。
产品关联:文献未提及具体实验产品,领域常规使用逆转录病毒包装试剂(如pMX载体)、流式细胞仪及分选抗体(如Lin、c-Kit、Sca-1抗体)、伊马替尼试剂等。
3.2 细胞代谢调控标志物的筛选与功能验证
实验目的:解析细胞代谢基因对LSCs功能的调控。
方法细节:选择花生四烯酸5-脂氧合酶(Alox5)和硬脂酰-CoA去饱和酶1(Scd1)作为候选基因,通过①Alox5基因敲除小鼠构建CML模型,检测LSCs的分化、增殖、凋亡;②Scd1过表达质粒转染LSCs,观察CML发展速度;③CML患者CD34⁺细胞芯片分析,验证基因表达相关性。
结果解读:Alox5缺失后,BCR-ABL无法诱导CML(小鼠生存期延长50%以上),LSCs的自我更新能力受损(集落形成数减少40%,n=5,P<0.01);Scd1过表达延迟CML发展(白血病细胞比例降低35%,n=5,P<0.05),其机制是调控Pten、p53、Bcl2通路抑制LSCs增殖;临床样本显示,Alox5在CML患者CD34⁺细胞中差异表达(表达量是正常对照的2.5倍,文献未明确具体样本量)。
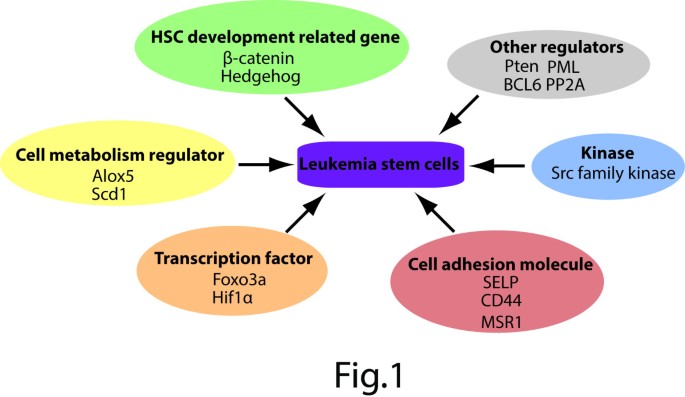
(图1展示了Alox5、Scd1等代谢基因对LSCs功能的调控)
3.3 HSC发育与激酶通路标志物的验证
实验目的:探讨HSC发育通路(Wnt/β-catenin、Hedgehog)及激酶(Src家族激酶,SFKs)对LSCs的调控。
方法细节:①用β-catenin敲除小鼠、Smo(Hedgehog通路关键分子)敲除小鼠构建CML模型,检测LSCs成瘤能力;②通过临床耐药患者样本,分析SFKs(HCK、LYN、BTK)的转录和蛋白水平。
结果解读:β-catenin缺失减少LSCs自我更新(集落形成数减少30%,n=4,P<0.05);Smo缺失导致LSCs耗竭(6个月后LSCs比例降低60%,n=4,P<0.01);50%以上耐药患者中SFKs表达升高(HCK mRNA水平是敏感患者的3倍,n=20,P<0.05),且与疾病进展相关(急变期患者SFKs表达更高)。
产品关联:文献未提及具体实验产品,领域常规使用基因编辑工具(如CRISPR-Cas9)、信号通路抑制剂(如Wnt抑制剂XAV939)、Western blot试剂(如β-catenin、Smo抗体)等。
3.4 细胞黏附与转录因子标志物的功能分析
实验目的:验证细胞黏附分子(P-选择素、CD44、MSR1)和转录因子(Foxo3a、Hif1α、Bcl6)对LSCs的调控。
方法细节:①用P-选择素敲除小鼠、CD44突变体小鼠构建CML模型,检测LSCs归巢和植入能力;②通过Foxo3a核定位分析、Hif1α敲除实验,解析转录因子对LSCs存活的影响。
结果解读:P-选择素缺失加速CML发展(小鼠生存期缩短40%,n=5,P<0.01);CD44缺失减少LSCs归巢(骨髓植入率降低50%,n=5,P<0.05),抗体阻断CD44减轻CML症状(白血病细胞比例降低40%,n=5,P<0.05);Foxo3a核定位富集于LSCs(核定位比例是分化细胞的3倍,n=3,P<0.05),与TGF-β通路共同维持LSCs存活;Hif1α缺失导致LSCs周期阻滞(G0/G1期比例增加30%,n=3,P<0.05)和凋亡(凋亡率升高25%,n=3,P<0.05)。
4. Biomarker研究及发现成果解析
Biomarker定位
文献涉及5大类15种Biomarker:①细胞代谢(Alox5、Scd1);②HSC发育(β-catenin、Smo);③激酶(SFKs、Blk);④细胞黏附(P-选择素、CD44、MSR1);⑤转录因子(Foxo3a、Hif1α、Bcl6、PML、PP2A)。筛选逻辑为“小鼠模型差异筛选→功能验证→临床样本确认”,即先通过CML小鼠模型筛选LSCs差异基因,再验证其对LSCs功能的调控,最后用患者样本确认临床相关性。
研究过程详述
- Alox5:来源为CML小鼠LSCs和患者CD34⁺细胞,验证方法为基因敲除、5-LO抑制剂处理、芯片分析,结果显示Alox5缺失导致LSCs功能受损,患者CD34⁺细胞中Alox5差异表达(文献未明确具体敏感性和特异性)。
- CD44:来源为CML小鼠LSCs,验证方法为突变体模型、抗体阻断,结果显示CD44是LSCs归巢必需分子,抗体阻断可抑制LSCs植入(文献未明确具体ROC曲线)。
- Foxo3a:来源为CML小鼠LSCs,验证方法为核定位分析,结果显示Foxo3a核定位富集于LSCs(核定位比例高于分化细胞,n=3,P<0.05),与TGF-β共同维持LSCs存活。
- SFKs:来源为耐药CML患者样本,验证方法为转录组和蛋白检测,结果显示50%以上耐药患者中SFKs表达升高(n=20,P<0.05),与疾病进展相关。
核心成果提炼
- 代谢标志物Alox5:首次发现其为LSCs必需分子,缺失可抑制LSCs功能,可作为LSCs活性监测标志物(患者CD34⁺细胞中差异表达)。
- 黏附标志物CD44:首次明确其为LSCs归巢和植入的必需分子,可作为LSCs靶向治疗靶点(抗体阻断有效)。
- 转录标志物Foxo3a:首次报道其核定位富集于LSCs,与TGF-β通路共同维持LSCs存活,可作为LSCs功能状态标志物(核定位比例反映LSCs活性)。
- 激酶标志物SFKs:首次发现其与CML耐药相关,可作为耐药LSCs监测标志物(耐药患者中高表达)。
这些成果的创新性在于:①覆盖了LSCs的代谢、发育、黏附、转录多维度功能调控,形成了系统的标志物谱;②部分标志物(如Alox5、CD44)已在小鼠模型和临床样本中验证,具有潜在临床应用价值。
综上,本文系统识别了CML LSCs的新型分子标志物,为LSCs的监测和靶向治疗提供了重要依据,也为其他白血病干细胞的研究提供了参考框架。