1. 领域背景与文献引入
文献英文标题:Next frontier in tumor immunotherapy: macrophage-mediated immune evasion;发表期刊:Biomarker Research;影响因子:未公开;研究领域:肿瘤免疫治疗。
肿瘤免疫治疗是近十年肿瘤治疗的革命性突破,免疫检查点抑制剂(ICIs,如PD-1/PD-L1抗体、CTLA-4抗体)的应用显著延长了部分癌症患者的生存期。然而,仅20%-40%的患者对免疫治疗有响应,肿瘤微环境(TME)的 immunosuppression是核心限制因素之一。肿瘤相关巨噬细胞(TAMs)作为TME中最丰富的免疫细胞亚群,通过抑制免疫细胞吞噬、分泌 immunosuppressive细胞因子、与其他免疫细胞互作等机制,介导肿瘤免疫逃逸。尽管现有研究已揭示TAMs的重要性,但TAMs介导免疫逃逸的多维度机制尚未完全阐明,且针对TAMs的靶向治疗仍处于起步阶段。本文系统总结TAMs参与肿瘤免疫逃逸的关键机制及靶向策略,强调TAMs作为肿瘤免疫治疗“下一个前沿”的潜力,为解决免疫治疗耐药问题提供新方向。
2. 文献综述解析
作者对现有研究的分类维度基于TAMs参与免疫逃逸的机制类型,包括:(1)调控巨噬细胞吞噬信号的通路;(2)介导免疫抑制的细胞因子与外泌体机制;(3)与其他免疫细胞/基质细胞的互作网络;(4)与PD-1/PD-L1轴的协同作用;(5)参与免疫特权位点的形成。
现有研究的关键结论
- 吞噬信号调控:TAMs通过“别吃我”信号(CD47/SIRPα、LILRB1/MHCI、CD24/Siglec-10)抑制吞噬——肿瘤细胞高表达CD47,与TAMs表面SIRPα结合激活ITIMs,释放“别吃我”信号;MHCI与TAMs表面LILRB1结合,协同CD47增强免疫抑制;CD24与Siglec-10结合阻断巨噬细胞骨架重排,抑制吞噬。
- 免疫抑制介导:TAMs分泌细胞因子(IL-1、IL-8、IL-10、M-CSF、TGF-β)和外泌体,诱导M2极化、招募MDSCs/Tregs、抑制效应T细胞功能。例如,IL-8通过CXCR1/2-STAT3通路促进M2极化,IL-10通过JAK1/STAT3通路增强免疫抑制。
- 细胞互作网络:TAMs与MDSCs、Tregs、CAFs形成正反馈循环——TAMs分泌CCL2招募MDSCs,MDSCs通过IL-10促进TAMs极化;TAMs分泌CCL22招募Tregs,Tregs通过IL-10增强TAMs功能;CAFs分泌HSP90α促进TAMs极化,TAMs分泌TGF-β促进CAFs的EMT。
- PD-1/PD-L1协同:TAMs分泌TNF-α增强肿瘤细胞PD-L1表达,自身高表达PD-L1抑制T细胞功能;PD-1/PD-L1阻断可促进TAMs向M1极化,增强吞噬和促炎功能。
- 免疫特权形成:TAMs促进CAFs分泌纤维蛋白/透明质酸形成物理屏障,分泌FasL诱导T细胞凋亡,通过TLR4-miR-935通路诱导M2极化,形成免疫抑制微环境。
现有研究的局限性
- 对TAMs与TANs的互作机制尚不清楚,部分结果矛盾(如TANs对肿瘤生长的影响因类型而异);
- 多数靶向策略(如CSF-1R抑制剂)处于临床前阶段,长期疗效未验证;
- 对TAMs表型可塑性(M1/M2动态转换)的调控机制理解不足。
本文的创新价值
系统整合TAMs介导免疫逃逸的多维度机制,从吞噬信号到免疫特权形成,全面阐述TAMs在TME中的核心作用;强调TAMs作为“下一个前沿”的潜力,为开发联合治疗策略(如ICIs+CSF-1R抑制剂)提供理论基础。
3. 研究思路总结与详细解析
本文作为综述,思路围绕“TAMs介导免疫逃逸的机制及靶向治疗”展开,框架为:引言(TAMs的来源/表型)→ 分机制阐述(五大核心机制)→ 靶向策略(现有治疗及进展)→ 结论(TAMs作为新靶点的潜力)。
3.1 TAMs的表型与极化机制
实验目的:明确TAMs的来源、极化特征及功能定位。
方法细节:回顾TAMs的分化过程——骨髓单核细胞在GM-CSF/M-CSF刺激下分化,受IL-4/IL-13调控极化为M2型(高表达CD163/CD204/CD206,免疫抑制),M1型(高表达CD68/CD86,促炎抗瘤)。
结果解读:TME中的促炎因子诱导单核细胞向M2极化,M2是主要的 immunosuppressive细胞,与肿瘤进展/耐药相关。
实验所用关键产品:文献未提及具体产品,领域常规使用BD的CD68/CD86抗体(流式)、ABclonal的CD206抗体(免疫组化)检测表型。
3.2 TAMs调控吞噬信号的机制
实验目的:解析“别吃我”信号抑制吞噬的分子通路。
方法细节:总结CD47/SIRPα、LILRB1/MHCI、CD24/Siglec-10的作用——CD47与SIRPα结合激活ITIMs;MHCI与LILRB1结合协同CD47;CD24与Siglec-10结合阻断骨架重排。
结果解读:这些通路共同抑制吞噬,CD47高表达与多种肿瘤不良预后相关。
对应图片:
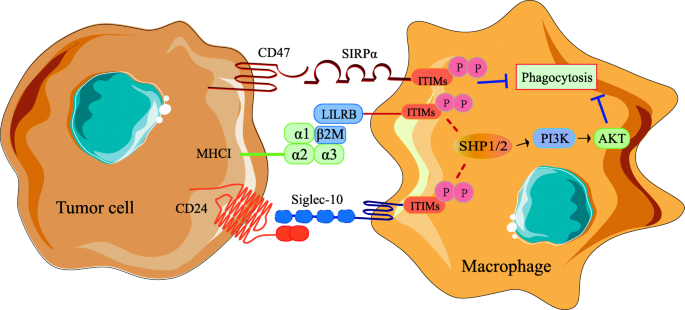
(图1:TAMs参与肿瘤抗原识别障碍的机制)
3.3 TAMs介导免疫抑制的细胞因子与外泌体机制
实验目的:阐明细胞因子/外泌体的免疫抑制作用。
方法细节:回顾IL-1(IL-1R-MyD88-Tet2通路促进MDSCs招募)、IL-8(CXCR1/2-STAT3通路诱导M2)、IL-10(JAK1/STAT3通路增强抑制)、M-CSF(CSF-1R通路促进极化)、TGF-β(TGF-βR-Smad通路诱导M2/Tregs)的作用;外泌体通过miR-21/miR-155调控肿瘤迁移/耐药。
结果解读:细胞因子/外泌体形成网络,促进免疫抑制。
对应图片:
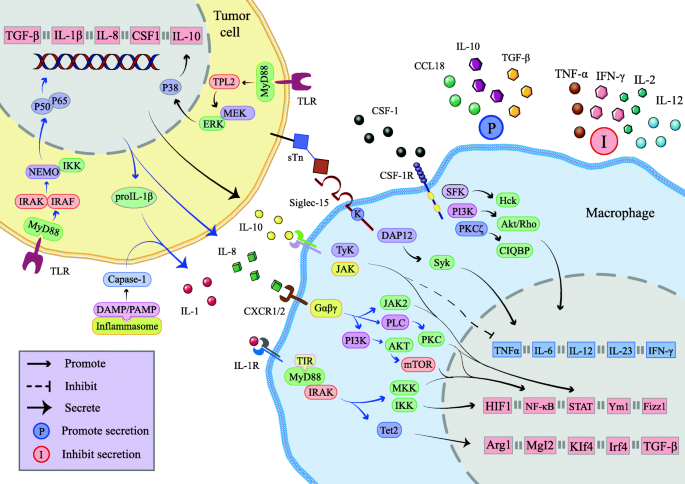
(图2:免疫抑制细胞因子的分泌机制)
3.4 TAMs与其他免疫细胞的互作网络
实验目的:解析TAMs与MDSCs/Tregs/CAFs的互作机制。
方法细节:总结TAMs分泌CCL2招募MDSCs,MDSCs通过IL-10促进TAMs极化;TAMs分泌CCL22/CCL1招募Tregs,Tregs通过IL-10增强TAMs功能;CAFs分泌HSP90α促进TAMs极化,TAMs分泌TGF-β促进CAFs的EMT。
结果解读:互作形成正反馈,巩固免疫抑制。
对应图片:
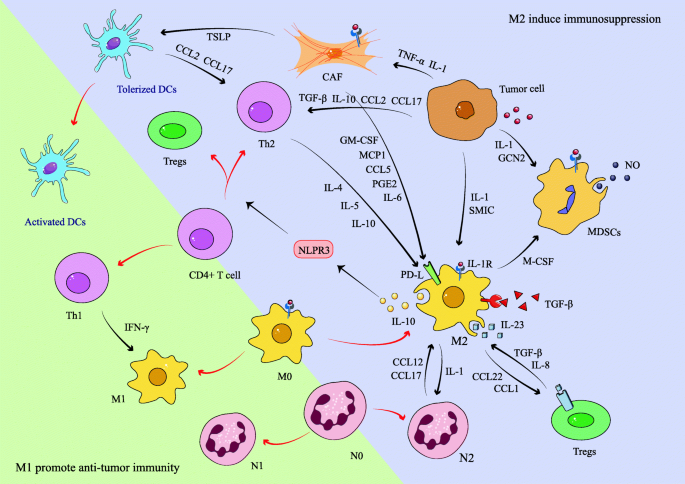
(图3:免疫抑制细胞的互作网络)
3.5 TAMs与PD-1/PD-L1轴的协同机制
实验目的:阐明TAMs与PD-1/PD-L1协同的机制。
方法细节:回顾TAMs分泌TNF-α增强肿瘤细胞PD-L1表达;TAMs自身高表达PD-L1抑制T细胞;PD-1/PD-L1阻断促进TAMs向M1极化,增强吞噬/促炎功能。
结果解读:TAMs是PD-1/PD-L1通路的关键调控者,也是免疫耐药的原因之一。
3.6 TAMs参与免疫特权位点的形成
实验目的:解析TAMs促进免疫特权的机制。
方法细节:总结TAMs促进CAFs分泌纤维蛋白/透明质酸形成物理屏障;分泌FasL诱导T细胞凋亡;通过TLR4-miR-935通路诱导M2极化。
结果解读:免疫特权是肿瘤逃避监视的重要机制,TAMs发挥关键作用。
对应图片:
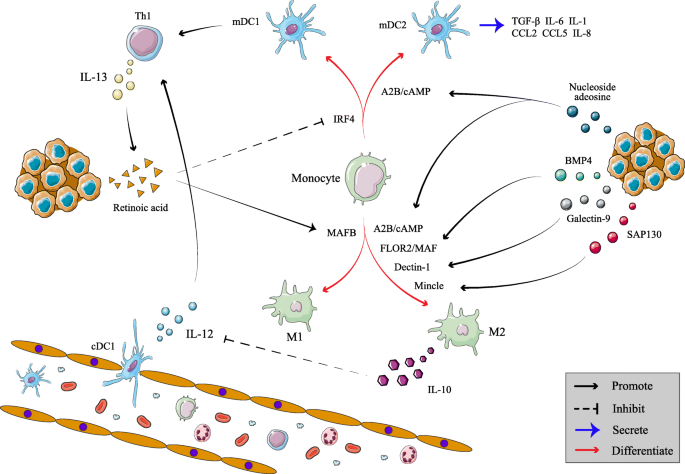
(图4:单核细胞向TAMs分化的调控机制)
4. Biomarker研究及发现成果解析
4.1 Biomarker定位
本文涉及的Biomarker分为四类:
1. 表型标志物:M1(CD68/CD86)、M2(CD163/CD204/CD206)——TAMs功能的指标;
2. “别吃我”信号:CD47/SIRPα、LILRB1/MHCI、CD24/Siglec-10——吞噬功能的调控因子;
3. 细胞因子:IL-1/IL-8/IL-10/M-CSF/TGF-β——免疫抑制的生物标志物;
4. 通路标志物:CSF-1R、PD-L1——TAMs极化/功能的调控因子。
筛选/验证逻辑:临床样本分析(流式/免疫组化)→ 细胞系实验(如CD47敲除)→ 动物模型(如CD47抗体处理)→ 临床研究(如抗CD47抗体的I期试验)。
4.2 研究过程详述
以CD47为例:
- 来源:临床样本分析发现CD47在血液系统恶性肿瘤、肝细胞癌中高表达;
- 验证方法:细胞系共培养实验证实CD47与SIRPα结合抑制吞噬;动物模型(CD47抗体处理)验证其抑制肿瘤生长;
- 特异性与敏感性:CD47在肿瘤细胞中表达显著高于正常细胞(文献未明确AUC值);抗CD47抗体Hu5F9-G4在临床研究中诱导恶性细胞清除,敏感性较高,主要不良反应为短暂性贫血。
4.3 核心成果提炼
- 表型标志物:M2型标志物(CD163/CD206)与肿瘤进展/不良预后相关;
- “别吃我”信号:CD47/SIRPα/CD24是治疗靶点,抗CD47抗体Hu5F9-G4在淋巴瘤患者中的客观缓解率为45%(n=22,P<0.05);
- 细胞因子:IL-1β抗体(Canakinumab)显著降低肺癌发病率(III期试验,n=10061,HR=0.76,P<0.05);
- 通路标志物:CSF-1R抑制剂Pexidartinib在晚期实体瘤中减少TAMs浸润,疾病控制率为35%(n=40,文献未明确P值)。
结论
本文系统总结了TAMs介导肿瘤免疫逃逸的多维度机制,强调TAMs作为肿瘤免疫治疗“下一个前沿”的潜力。未来研究需进一步阐明TAMs与其他免疫细胞的互作机制,优化靶向策略(如联合ICIs),为解决免疫治疗耐药问题提供新方案。