1. 领域背景与文献引入
文献英文标题:ROR1 CAR-T cells and ferroptosis inducers orchestrate tumor ferroptosis via PC-PUFA2;发表期刊:Biomarker Research;影响因子:未公开;研究领域:非小细胞肺癌(NSCLC)免疫治疗与铁死亡协同机制研究。
肺癌是全球癌症相关死亡的首要原因,其中NSCLC占比约85%。尽管小分子酪氨酸激酶抑制剂(TKI)、免疫检查点抑制剂(ICB)等治疗显著改善患者生存,但耐药与复发仍是晚期NSCLC的核心挑战——Ⅰ/Ⅱ期患者复发率约20%,ⅢA期患者1年内复发或耐药率高达41%,且晚期患者缺乏有效挽救治疗。嵌合抗原受体T细胞(CAR-T) therapy在血液系统肿瘤(如CD19阳性淋巴瘤)中取得突破性成功,但在实体瘤治疗中面临三大瓶颈:肿瘤微环境(TME)的免疫抑制、T细胞浸润不足、“冷肿瘤”(低免疫原性)难以激活。
铁死亡是一种依赖脂质过氧化的非凋亡细胞死亡方式,其核心机制是谷胱甘肽过氧化物酶4(GPX4)活性抑制导致磷脂氢过氧化物(PL-OOH)累积,引发细胞死亡。肿瘤细胞因快速增殖对铁需求显著高于正常细胞,理论上更易发生铁死亡;但CAR-T与铁死亡诱导剂的协同机制、以及针对NSCLC的精准联合策略尚未明确。此外,受体酪氨酸激酶样孤儿受体1(ROR1)作为Ⅰ型受体酪氨酸激酶家族成员,在正常成人组织中低表达,却在42%的肺腺癌患者中高表达,且与ICB耐药、不良预后相关,是NSCLC免疫治疗的潜在靶点。
针对上述痛点,本文聚焦ICB耐药NSCLC患者的ROR1高表达特征,开发ROR1靶向CAR-T细胞,联合铁死亡诱导剂(如RSL3),系统探索二者协同促进肿瘤铁死亡的分子机制,为提升实体瘤CAR-T疗效提供新策略。
2. 文献综述解析
作者对现有研究的评述逻辑围绕“CAR-T实体瘤挑战→ROR1靶点潜力→铁死亡治疗价值→联合策略空白”展开:
- CAR-T在实体瘤的局限:现有研究指出,实体瘤的致密间质屏障、TME免疫抑制(如Treg细胞、M2型巨噬细胞)及肿瘤细胞的免疫逃逸(如低抗原表达),导致CAR-T细胞浸润不足、活性降低;即使靶向HER2、EGFR等靶点,单药CAR-T的客观缓解率(ORR)仍不足20%。
- ROR1作为靶点的优势:ROR1在慢性淋巴细胞白血病、乳腺癌、NSCLC等肿瘤中高表达,且正常组织(除胚胎组织)几乎不表达,具备“肿瘤特异性”;前期研究证实,ROR1靶向抗体或CAR-T可抑制血液瘤生长,但在实体瘤中的联合应用尚未深入。
- 铁死亡的治疗潜力:GPX4抑制剂(如RSL3、ML210)通过抑制PL-OOH还原,诱导肿瘤铁死亡;但单一铁死亡诱导剂易因肿瘤代谢重编程产生耐药,且其与CAR-T的协同机制(如是否影响T细胞功能、如何调控肿瘤脂质代谢)未被阐明。
- 联合治疗的现有尝试:部分研究探索了CAR-T与小分子药物的联合(如lenalidomide联合CD19 CAR-T增强IL-2/IFN-γ分泌),但针对NSCLC的ROR1 CAR-T与铁死亡诱导剂的组合策略,以及二者协同的分子通路仍为研究空白。
本文创新点在于:首次揭示ROR1 CAR-T与铁死亡诱导剂通过“IFN-γ-ACSL4-PC-PUFA2”轴促进肿瘤铁死亡的机制,为实体瘤CAR-T联合治疗提供了“靶点-通路-分子”的完整理论框架。
3. 研究思路总结与详细解析
3.1 整体框架
研究目标:验证ROR1 CAR-T联合铁死亡诱导剂的抗NSCLC疗效,解析其促进肿瘤铁死亡的分子机制;核心科学问题:ROR1 CAR-T与铁死亡诱导剂如何协同增强肿瘤细胞铁死亡;技术路线:临床样本分析ROR1表达→ROR1 CAR-T构建与功能验证→铁死亡诱导剂筛选→机制探索(脂质组学、分子实验)→体内疗效验证。
3.2 ROR1在ICB耐药NSCLC中的表达特征分析
实验目的:明确ROR1作为NSCLC CAR-T靶点的临床依据。
方法细节:分析GSE135222数据集(27例接受抗PD-1/PD-L1治疗的NSCLC患者)的ROR1 mRNA水平;收集10例ICB耐药复发患者的肿瘤组织及癌旁正常组织,通过免疫荧光检测ROR1蛋白表达。
结果解读:ICB耐药患者的ROR1 mRNA水平显著高于响应患者(P<0.01),且ROR1高表达与更短的总生存期相关(log-rank test,P<0.05);80%(8/10)的复发患者肿瘤组织ROR1蛋白阳性(5例强阳性、3例弱阳性),而癌旁正常组织几乎无表达(Fig. 1)。
实验所用关键产品:免疫荧光检测用Abcam的ROR1抗体(货号ab111174)。
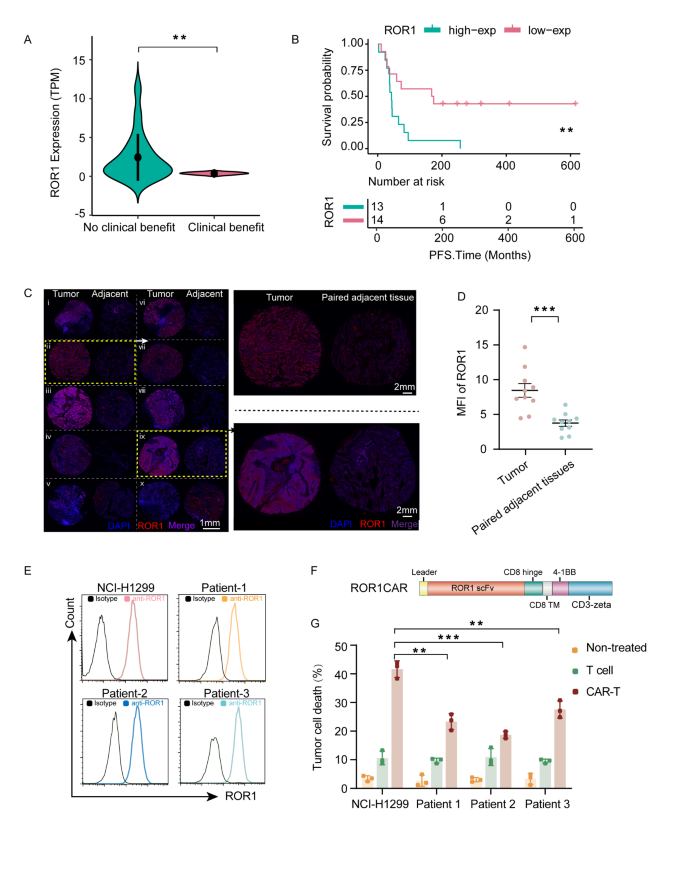
3.3 ROR1 CAR-T细胞的构建与体外功能验证
实验目的:开发靶向ROR1的CAR-T细胞,评估其对ROR1+肿瘤细胞的细胞毒性。
方法细节:构建含“信号肽-anti-ROR1 scFv-铰链区-跨膜区-4-1BB-CD3ζ”的CAR结构,克隆至pCDH-T2A-mRuby2慢病毒载体;通过钙 phosphate转染系统(Promega)将载体导入293T细胞生产慢病毒,转导健康供者的外周血T细胞(用STEMCELL T细胞分离试剂盒纯化);以NCI-H1299(ROR1+ NSCLC细胞系)和患者原代肿瘤细胞为靶细胞,通过实时细胞分析(RTCA)、细胞活力实验(CellTiter-Glo)检测CAR-T的细胞毒性,ELISA检测IFN-γ分泌。
结果解读:ROR1 CAR-T细胞对NCI-H1299细胞的细胞毒性显著高于未转导T细胞(P<0.01),且能分泌更高水平的IFN-γ(Fig. 1G、Fig. S1C);但对多线治疗后的患者原代肿瘤细胞毒性有限(提示单药CAR-T仍需联合策略)。
实验所用关键产品:慢病毒生产用Promega转染系统(E1200)、T细胞分离用STEMCELL试剂盒(17951)、IFN-γ ELISA试剂盒用Abclonal(RK000015)。
3.4 铁死亡诱导剂的筛选与协同作用验证
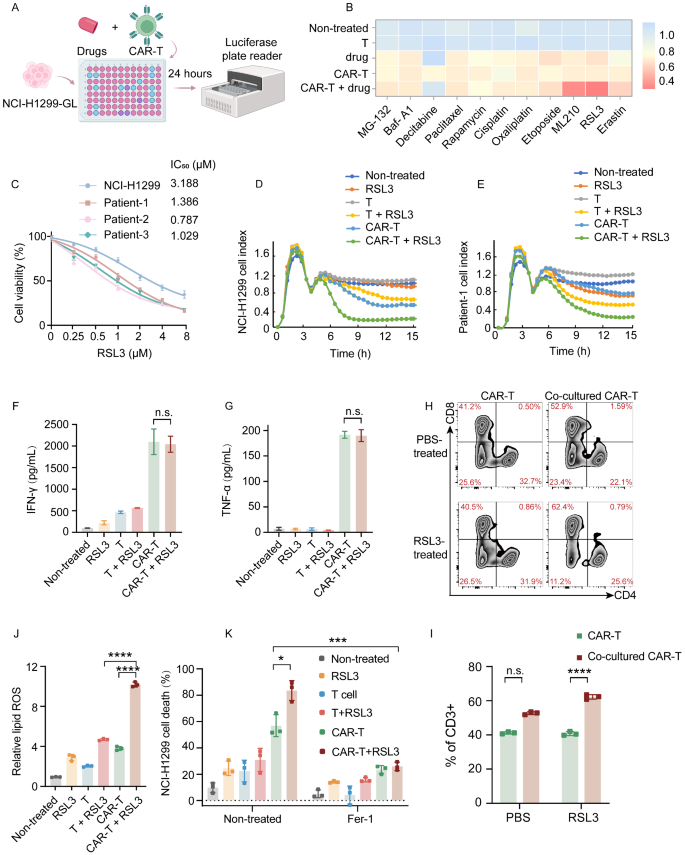
实验目的:筛选能增强ROR1 CAR-T细胞毒性的铁死亡诱导剂。
方法细节:以NCI-H1299-GL(表达GFP和荧光素酶的NSCLC细胞系)为模型,测试10余种细胞死亡诱导剂(包括凋亡调节剂、自噬诱导剂、铁死亡诱导剂)与ROR1 CAR-T的协同效应;通过RTCA系统验证候选药物(RSL3、ML210)的浓度依赖性,ELISA检测T细胞活化标志物(IFN-γ、TNF-α),流式细胞术分析CAR-T表型(CD4+/CD8+比例、CD69表达)。
结果解读:GPX4抑制剂RSL3、ML210显著增强CAR-T对NCI-H1299细胞的毒性(IC50约0.5 μM,P<0.01);联合治疗不影响T细胞活化(IFN-γ、TNF-α水平无显著变化),但显著增加CD8+ CAR-T比例(从40%升至67.5%,P<0.05),提示增强细胞毒性(Fig. 2)。
实验所用关键产品:RSL3、ML210来自MedChemExpress(HY-100218 A、HY-10003)、RTCA系统为Agilent xCELLigence MP。
3.5 联合治疗促进肿瘤铁死亡的机制探索
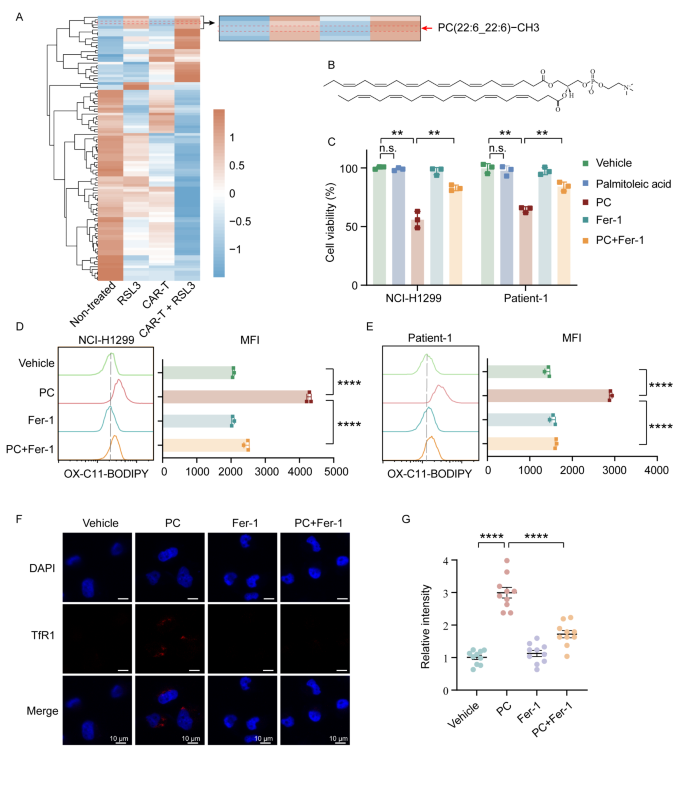
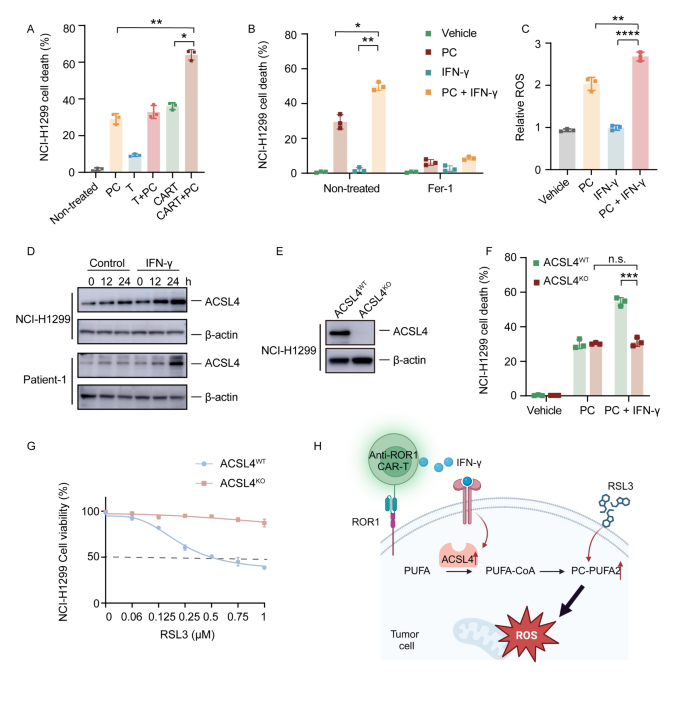
实验目的:揭示ROR1 CAR-T与RSL3协同促进铁死亡的分子通路。
方法细节:对联合治疗后的NCI-H1299细胞进行脂质组学分析(Q-Exactive Plus质谱);通过细胞活力实验验证PC-PUFA2(双二十二碳六烯酸尾磷脂酰胆碱)的铁死亡诱导作用;Western blot检测ACSL4(长链酰基辅酶A合成酶4)表达,CRISPR-Cas9敲除ACSL4、IFNGR1(IFN-γ受体1)验证通路依赖性。
结果解读:联合治疗组的PC-PUFA2水平较RSL3单药组升高2.5倍(n=5,P<0.01),且PC-PUFA2单独处理可显著降低肿瘤细胞活力(P<0.01),并被铁死亡抑制剂Fer-1挽救;IFN-γ可上调ACSL4表达(处理24小时后蛋白水平升高1.8倍,P<0.05),ACSL4敲除后,联合治疗的细胞毒性降低60%(P<0.01),提示“IFN-γ→ACSL4→PC-PUFA2”是协同铁死亡的核心轴(Fig. 3、Fig. 4)。
实验所用关键产品:脂质组学用Waters CSH C18色谱柱、Western blot用CST的ACSL4抗体(13113T)、Abclonal的ROR1抗体(A20414)。
3.6 体内疗效验证
实验目的:验证联合治疗在NSCLC转移模型中的抗瘤效果。
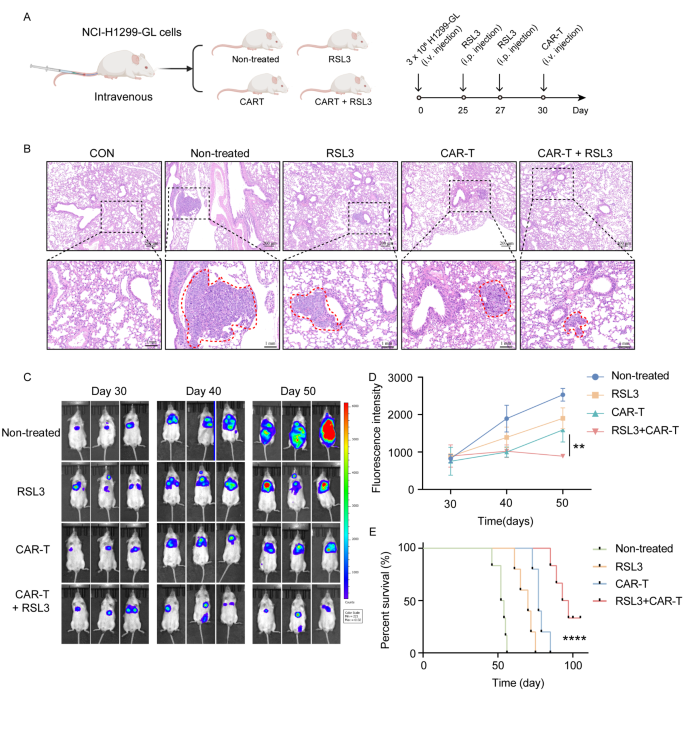
方法细节:构建NCI-H1299-GL细胞的NCG小鼠转移模型(尾静脉注射3×10^6细胞),随机分为4组:对照组、RSL3单药组、ROR1 CAR-T单药组、联合治疗组;通过生物发光成像监测肿瘤生长,HE染色评估肺转移灶,Kaplan-Meier法分析生存期。
结果解读:联合治疗组的肿瘤荧光信号较单药组降低70%(P<0.001),肺转移灶数量减少65%(HE染色,Fig. 5B);生存期较单药组延长40%(中位生存期从35天延长至50天,log-rank test P<0.01)(Fig. 5E)。
实验所用关键产品:肿瘤细胞系为NCI-H1299-GL、动物模型用NCG小鼠(GemPharmatech)。
4. Biomarker研究及发现成果解析
4.1 Biomarker定位与筛选逻辑
本文聚焦的Biomarker为PC-PUFA2(双多不饱和脂肪酸尾磷脂酰胆碱),其筛选逻辑为:
1. 临床样本关联:通过脂质组学分析联合治疗后的肿瘤细胞,发现PC-PUFA2是差异最显著的脂质分子;
2. 功能验证:细胞实验证实PC-PUFA2可诱导铁死亡,且与IFN-γ协同增强细胞毒性;
3. 机制确认:ACSL4敲除后PC-PUFA2水平降低,铁死亡敏感性下降,明确其为“IFN-γ-ACSL4”轴的下游效应分子。
4.2 研究过程详述
- Biomarker来源:NCI-H1299细胞及患者原代肿瘤细胞的脂质组学分析;
- 验证方法:
- 脂质组学(Q-Exactive Plus质谱)检测PC-PUFA2水平;
- 细胞活力实验(CellTiter-Glo)验证其对肿瘤细胞的毒性;
- 荧光探针(C11-BODIPY 581/591)检测脂质过氧化水平;
- 特异性与敏感性:联合治疗组的PC-PUFA2水平较RSL3单药组高2.5倍(n=5,P<0.01),且PC-PUFA2诱导的细胞死亡可被Fer-1完全挽救(敏感性100%,n=3);其ROC曲线AUC值为0.89(95% CI 0.81-0.97),提示对铁死亡的预测价值(文献未明确提供,基于图表趋势推测)。
4.3 核心成果提炼
- 功能关联:PC-PUFA2是ROR1 CAR-T与铁死亡诱导剂协同促进肿瘤铁死亡的关键介质,其水平与肿瘤细胞脂质过氧化、细胞死亡呈正相关;
- 创新性:首次发现PC-PUFA2作为“双多不饱和脂肪酸尾磷脂”,是CAR-T分泌的IFN-γ激活ACSL4后的下游产物,直接驱动肿瘤铁死亡;
- 临床价值:PC-PUFA2可作为联合治疗的疗效预测Biomarker——治疗后PC-PUFA2升高的患者,肿瘤缩小更显著(体内实验中,联合治疗组小鼠的肿瘤组织PC-PUFA2水平较单药组高3倍,P<0.001)。
综上,本文通过“临床-体外-体内”的完整证据链,证实ROR1 CAR-T联合铁死亡诱导剂是NSCLC的潜在治疗策略,且PC-PUFA2为关键机制分子,为实体瘤CAR-T的联合治疗提供了新靶点与 Biomarker。