1. 领域背景与文献引入
文献英文标题:Machine learning and systems genomics approaches for multi-omics data;发表期刊:Biomarker Research;影响因子:未公开;研究领域:生物信息学-多组学数据整合与系统基因组学
领域共识:精准医学的核心是通过个体的遗传、分子特征实现个性化医疗决策,多组学数据整合是精准医学发展的关键技术支撑。自2015年美国正式提出精准医学概念以来,领域内已取得多项核心技术突破,包括高通量多组学检测技术的普及、经典机器学习算法在生物信息学中的应用等。当前研究热点聚焦于如何通过高效的计算方法整合多组学数据,以揭示疾病发生发展的分子机制、筛选特异性生物标志物,但现有多组学整合方法缺乏系统的框架梳理,不同方法的适用场景与优缺点未得到清晰的对比总结,这为临床转化与后续研究带来了技术瓶颈。本文正是针对这一空白,系统综述了机器学习与系统基因组学(MLSG)领域的软件框架,为多组学数据的整合分析提供全面的技术参考与未来方向指引。
2. 文献综述解析
本文作者以多组学数据的整合策略为分类维度,将现有机学习与系统基因组学方法划分为模型基整合(MBI)、串联基整合(CBI)、转换基整合(TBI)三大类,系统梳理了各类方法的技术原理、应用场景、优势与局限性,并结合数据降维与特征选择方法,构建了完整的MLSG多组学分析技术体系。
现有研究已证实,多组学数据整合分析相比单一组学分析,能更全面地解析表型与基因型的关联,例如在肿瘤亚型分类、药物反应预测等领域展现出更高的准确性与敏感性。从技术方法来看,模型基整合方法的优势在于能够合并不同数据类型的预测模型,且可基于同一表型的不同患者数据集进行模型构建;串联基整合方法的优势在于流程相对简单,可适配多种经典机器学习算法;转换基整合方法的优势在于能够整合连续型与分类型等不同类型的多组学数据。但现有方法也存在明显局限性,模型基整合方法的模型构建过程复杂,对计算资源要求较高;串联基整合方法易受高维数据的影响,存在过拟合风险;转换基整合方法对中间转换形式(如核矩阵、图结构)的选择依赖较大,缺乏标准化的转换策略。
本文的创新价值在于首次系统地将MLSG多组学整合方法划分为三大类,并对每类方法的具体实现框架、已有的研究应用案例进行了全面总结,同时补充了数据降维与特征选择方法的适配策略,填补了领域内对MLSG方法体系化梳理的空白,为不同研究场景下的多组学数据分析提供了清晰的技术选择路径。
3. 研究思路总结与详细解析
本文的研究目标是系统综述机器学习与系统基因组学领域的软件框架,明确各类多组学数据整合方法的技术逻辑、优势与局限性,并展望该领域的未来发展方向;核心科学问题是如何通过MLSG方法有效整合多组学数据,以更精准地解析表型与基因型的关联;技术路线遵循“分类综述整合框架→补充特征选择方法→总结未来方向”的逻辑闭环,全面覆盖了MLSG多组学分析的核心技术环节。
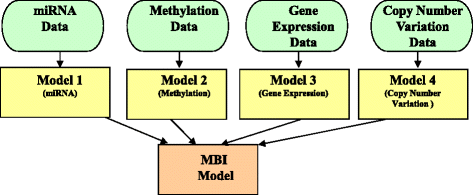
3.1 模型基整合(MBI)框架解析
本环节的核心目标是解析模型基整合方法的技术原理与应用场景。模型基整合方法的流程为:首先以不同类型的多组学数据为训练集生成多个独立模型,再将这些模型整合为最终的预测模型。具体实现框架包括多数投票法、集成分类器法、概率因果网络等,其中概率因果网络可通过贝叶斯网络整合高度异质性的数据,通过蒙特卡洛马尔可夫链模拟筛选最优网络结构,并利用贝叶斯信息准则评分避免过拟合。在应用案例中,该方法可整合微小RNA(miRNA)、甲基化、基因表达、拷贝数变异等多组学数据,构建卵巢癌生存预测的多维度模型,对比单一组学模型,整合模型的预测准确性得到显著提升。
文献未提及具体实验产品,领域常规使用Python/R语言的机器学习库(如scikit-learn、PyMC3)、生物信息学分析工具(如Cytoscape)等。
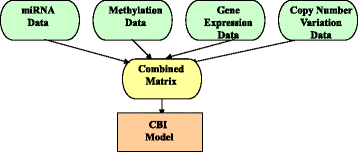
3.2 串联基整合(CBI)框架解析
本环节的核心目标是解析串联基整合方法的技术原理与应用场景。串联基整合方法的流程为:首先将多个多组学数据矩阵合并为一个大型输入矩阵,再利用经典机器学习算法进行分析。具体实现框架包括贝叶斯网络、多变量Cox LASSO模型、iCluster等,其中iCluster框架可同时完成数据整合与降维,将多组学数据合并为单一矩阵后进行肿瘤亚型分析。在应用案例中,Fridley等通过整合mRNA表达与单核苷酸多态性(SNP)数据,构建贝叶斯整合模型预测吉西他滨的癌症治疗反应,结果显示该模型相比单一数据类型分析具有更高的敏感性;Shen等利用iCluster框架整合胶质母细胞瘤的拷贝数、mRNA表达、甲基化数据,成功识别出3种不同的整合肿瘤亚型。
文献未提及具体实验产品,领域常规使用Python/R语言的机器学习库(如scikit-learn、glmnet)、生物信息学分析工具(如TCGAbiolinks)等。
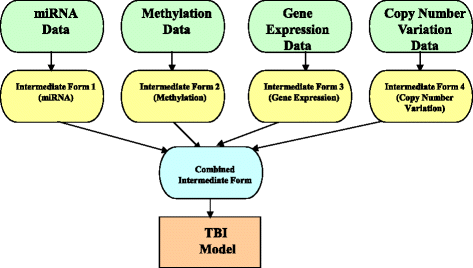
3.3 转换基整合(TBI)框架解析
本环节的核心目标是解析转换基整合方法的技术原理与应用场景。转换基整合方法的流程为:首先将每个多组学数据集转换为中间形式(如图结构、核矩阵),再将多个中间形式合并后构建模型。具体实现框架包括基于核的整合方法、基于图的半监督学习方法等。在应用案例中,Wahl等利用加权相关网络方法,将血清代谢组与全血基因表达数据转换为模块网络,构建部分相关网络分析体重变化的代谢与转录组关联,结果显示4个代谢物模块与2个基因表达模块与体重变化显著相关,揭示了长期体重变化与血清代谢物浓度的关联。
文献未提及具体实验产品,领域常规使用Python/R语言的机器学习库(如scikit-learn、igraph)、生物信息学分析工具(如WGCNA)等。
3.4 数据降维与特征选择方法解析
本环节的核心目标是解析与MLSG框架适配的数据降维与特征选择方法,以解决多组学数据的高维性问题。数据降维与特征选择方法分为外在方法与内在方法,外在方法如Biofilter可利用公共数据库的先验知识进行特征筛选;内在方法包括因子分析、ReliefF、卡方检验、主成分分析、遗传算法等。此外,还有混合方法(如信息增益与卡方检验结合)、基于包装器的特征选择方法(如最佳优先搜索策略),可进一步提升特征筛选的准确性与效率,降低模型过拟合风险。
文献未提及具体实验产品,领域常规使用Python/R语言的机器学习库(如scikit-learn、mlr)、生物信息学分析工具(如Biofilter)等。
4. Biomarker研究及发现成果
本文作为综述类文献,未直接发现新的生物标志物(Biomarker),但系统梳理了机器学习与系统基因组学方法在Biomarker筛选与验证中的应用逻辑,其核心流程为“多组学数据整合→特征选择筛选候选标记→模型验证与临床关联分析”,为精准医学领域的Biomarker开发提供了全面的技术路线参考。
从Biomarker的来源来看,涵盖了基因组(单核苷酸多态性、拷贝数变异)、转录组(基因表达)、表观基因组(甲基化)、代谢组(代谢物)等多组学层面的分子标记;验证方法包括基于机器学习模型的预测分析、临床样本的关联分析等。在特异性与敏感性方面,多组学整合方法相比单一组学方法,能显著提升Biomarker的敏感性,例如Fridley等的研究显示,贝叶斯整合模型在检测吉西他滨的基因组效应时,敏感性高于传统单一数据类型分析;Shen等的研究显示,iCluster框架整合多组学数据识别的肿瘤亚型,具有明确的分子特征与临床关联。
本文的核心成果在于总结了MLSG方法在Biomarker研究中的三大优势:一是能够整合多组学数据,发现单一组学无法识别的协同Biomarker;二是通过特征选择方法降低高维数据的干扰,提升Biomarker的特异性;三是可构建预测模型,验证Biomarker与表型(如疾病预后、药物反应)的关联。其创新性在于首次系统地将三类MLSG整合方法与Biomarker研究的应用场景进行对应,为不同研究需求下的Biomarker筛选提供了清晰的技术选择依据,例如模型基整合方法适用于多数据集的Biomarker整合验证,串联基整合方法适用于肿瘤亚型相关Biomarker的筛选,转换基整合方法适用于不同类型多组学数据的Biomarker关联分析。文献未提供具体的统计学数据(如ROC曲线AUC值、风险比HR),相关结果基于研究案例的定性描述。