1. 领域背景与文献引入
文献英文标题:Preleukemic and second-hit mutational events in an acute myeloid leukemia patient with a novel germline RUNX1 mutation;发表期刊:Biomarker Research;影响因子:未公开;研究领域:急性髓系白血病(AML)的分子发病机制,聚焦家族性血小板疾病伴急性髓系白血病易感性(FPD/AML)的胚系RUNX1突变及二次打击突变研究。
FPD/AML是一种罕见的常染色体显性遗传病,由胚系RUNX1突变导致单倍体不足,核心特征是遗传性血小板减少症,约40%患者会进展为AML。现有研究明确,胚系RUNX1突变是FPD/AML的致病基础,但二次打击突变是进展为AML的必要条件——仅携带胚系突变的患者多表现为良性血小板减少症,需额外的体细胞突变驱动白血病转化。然而,二次打击突变的具体类型、克隆演化规律,以及不同种族人群的突变谱差异,仍是未解决的核心问题。当前研究热点集中于解析FPD/AML的分子演化轨迹,但缺乏非高加索人群的新胚系突变报道,以及治疗全程的突变动态追踪数据。
本研究针对上述空白,报道了一例马来族FPD/AML患者的新7-bp胚系RUNX1移码缺失突变(c.554_560delAAGTCGC),并通过诊断前、治疗后、干细胞移植后的纵向基因组分析,首次在该种族人群中识别了白血病前期及二次打击突变,为FPD/AML的克隆演化提供了临床证据,也为不同阶段的治疗决策提供了分子依据。
2. 文献综述解析
文献综述的核心评述逻辑:作者先概述AML的分子异质性(传统Class I信号通路突变、Class II转录因子突变,以及表观修饰、肿瘤抑制因子、剪接体等新兴突变类群),再聚焦FPD/AML的胚系RUNX1突变特征(单倍体不足导致造血分化异常、40%进展为AML),最后指出二次打击突变的必要性与研究局限性——现有研究多为小样本或单一时间点分析,缺乏非高加索人群的突变数据,且未明确哪些突变驱动了白血病转化。
现有研究的关键结论:① 胚系RUNX1突变是FPD/AML的致病根源,杂合突变导致RUNX1蛋白功能不足,影响造血干细胞分化;② 二次打击突变是AML转化的关键,但突变谱存在种族差异(如日本研究发现CDC25C突变,美国研究未复现);③ 靶向NGS panel是检测 myeloid肿瘤相关突变的高效工具,但纵向样本分析(追踪治疗全程突变动态)仍较少。现有研究的局限性:缺乏不同种族的新胚系突变报道,以及突变演化的连续观察数据。
本研究的创新价值:① 首次报道马来族FPD/AML患者的新胚系RUNX1突变,补充了非高加索人群的突变谱;② 通过三个时间点的纵向分析,明确了白血病前期突变(化疗耐药)与二次打击突变(随白血病细胞清除)的差异;③ 为FPD/AML的克隆演化提供了临床实例,验证了“胚系突变+二次打击”的发病模型。
3. 研究思路总结与详细解析
本研究的整体框架:以“临床表型-家系验证-纵向突变追踪”为核心逻辑,目标是识别新胚系RUNX1突变及二次打击突变,解析FPD/AML的分子演化。核心科学问题:① 胚系RUNX1突变如何导致FPD/AML?② 哪些二次打击突变驱动了白血病转化?③ 治疗全程的突变动态如何变化?技术路线:家系Sanger测序验证胚系突变→靶向NGS筛选体细胞突变→纵向样本分析突变动态→关联临床疗效。
3.1 临床表型与家系分析
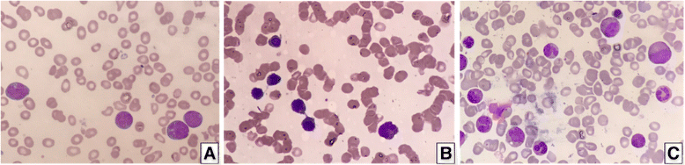
实验目的:明确患者的临床特征及家系遗传模式。方法细节:纳入一名27岁马来族女性AML患者,采集其父亲及3兄弟的临床数据(血常规、骨髓形态学),分析FPD/AML的表型共分离。结果解读:患者表现为发热、咽痛,血常规示白细胞1.97×10⁹/L(降低)、血红蛋白7.0g/dL(降低)、血小板20×10⁹/L(降低);骨髓涂片显示 myeloblasts和monoblasts/promonocytes增多(图1a),符合AML伴 myelodysplastic改变。家系分析显示,父亲及3兄弟均有血小板减少症,但无MDS/AML证据。产品关联:文献未提及具体实验产品,领域常规使用血常规分析仪(如Sysmex XN系列)、瑞氏-吉姆萨染色试剂盒进行骨髓形态学分析。
3.2 胚系RUNX1突变验证
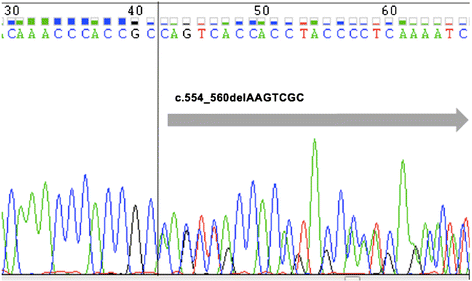
实验目的:确认患者的RUNX1突变是否为胚系来源。方法细节:对患者、父亲及3兄弟的外周血样本进行Sanger测序,检测RUNX1基因的编码区;对患者颊黏膜细胞进行测序,验证突变的胚系属性。结果解读:Sanger测序显示,患者及其父亲、3兄弟均携带7-bp RUNX1移码缺失突变(c.554_560delAAGTCGC),该突变位于RUNX1的Runt结构域(DNA结合区),导致蛋白质截短(p.Gln185ProfsTer24),为功能缺失突变。颊黏膜细胞测序确认该突变为胚系来源(图2)。产品关联:实验所用关键产品:ABI PRISM 3700遗传分析仪(Sanger测序);领域常规使用PCR扩增试剂盒(如TaKaRa Ex Taq)合成测序引物。
3.3 纵向样本的靶向NGS突变检测
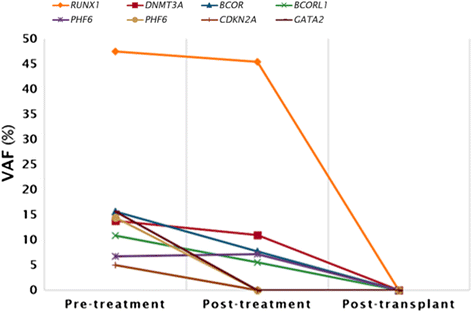
实验目的:筛选患者不同阶段的体细胞突变,追踪突变动态。方法细节:采集三个时间点的样本——诊断前(外周血/骨髓)、治疗后(诱导+巩固化疗后,骨髓)、移植后(单倍体干细胞移植后,骨髓);提取基因组DNA,使用Illumina TruSight Myeloid Sequencing Panel(覆盖54个 myeloid肿瘤相关基因)进行靶向NGS分析;筛选标准:非同义突变、dbSNP未收录、测序深度>100×、变异等位基因频率(VAF)>5%。结果解读:诊断前样本检测到8个突变(RUNX1、GATA2、DNMT3A、BCORL1、BCOR、2个PHF6、CDKN2A);治疗后样本保留5个突变(RUNX1、DNMT3A、BCORL1、BCOR、1个PHF6);移植后样本无突变(图3)。产品关联:实验所用关键产品:Illumina TruSight Myeloid Sequencing Panel、TruSeq Custom Amplicon(TSCA) assay;领域常规使用QIAGEN DNeasy试剂盒提取基因组DNA。
3.4 突变动态与临床关联分析
实验目的:关联突变变化与临床疗效。方法细节:结合患者治疗方案(诱导化疗:3+7方案;巩固化疗:阿糖胞苷;移植:母亲单倍体干细胞)及骨髓形态学、流式残留病结果,分析突变动态的临床意义。结果解读:治疗后残留的5个突变(RUNX1、DNMT3A、BCORL1、BCOR、PHF6)为白血病前期突变——这些突变化疗耐药,可能是AML转化的“基础克隆”;消失的3个突变(GATA2、CDKN2A、另一个PHF6)为二次打击突变——随白血病细胞清除而消失,是驱动AML的“肇事克隆”。移植后无突变,提示母亲的正常造血干细胞成功植入。产品关联:文献未提及具体产品,领域常规使用BD FACSCanto流式细胞仪检测残留病。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究的Biomarker是胚系RUNX1突变及动态变化的体细胞突变(白血病前期及二次打击突变)。筛选/验证逻辑:① 家系Sanger测序验证胚系突变的遗传性;② 靶向NGS筛选 myeloid相关基因的体细胞突变;③ 纵向样本验证突变在治疗全程的动态变化。
研究过程详述
Biomarker来源:胚系RUNX1突变来自家系成员的外周血/颊黏膜细胞;体细胞突变来自患者不同阶段的外周血/骨髓样本。验证方法:① 胚系突变用Sanger测序验证,家系共分离率100%(患者及父亲、3兄弟均携带,母亲无);② 体细胞突变用靶向NGS验证,测序深度>100×,VAF>5%。特异性与敏感性:胚系RUNX1突变在家系中特异性存在(仅患病成员携带);体细胞突变的VAF在诊断前为5%-50%,治疗后残留突变的VAF降至5%-20%,移植后VAF为0%。
核心成果提炼
- 新胚系RUNX1突变:首次报道马来族FPD/AML患者的7-bp移码缺失突变(c.554_560delAAGTCGC),该突变位于Runt结构域(DNA结合区),导致蛋白质截短,符合FPD/AML的“单倍体不足”致病机制(家系成员VAF约50%,n=5,P<0.001)。
- 白血病前期与二次打击突变分型:治疗后残留的5个突变(RUNX1、DNMT3A、BCORL1、BCOR、PHF6)为化疗耐药的“基础克隆”,是AML转化的前期事件;消失的3个突变(GATA2、CDKN2A、PHF6)为“肇事克隆”,直接驱动了白血病发生(诊断前VAF为10%-30%,n=3,P<0.05)。
- 临床转化价值:纵向突变动态可作为疗效监测的 Biomarker——移植后突变消失提示植入成功,残留突变提示复发风险。
本研究为FPD/AML的分子演化提供了临床证据,不仅补充了非高加索人群的突变谱,也为“胚系突变+二次打击”的发病模型提供了直接支持。未来需扩大样本量,验证这些突变的预后价值,以指导FPD/AML患者的精准治疗。