1. 领域背景与文献引入
文献英文标题:The orphan nuclear receptor EAR-2 (NR2F6) inhibits hematopoietic cell differentiation and induces myeloid dysplasia in vivo;发表期刊:Biomarker Research;影响因子:未公开;研究领域:骨髓增生异常综合征(MDS)发病机制与动物模型构建。
骨髓增生异常综合征(MDS)是起源于造血干细胞的恶性克隆性疾病,其核心矛盾在于:骨髓细胞的凋亡率显著增加,但恶性克隆仍能排挤正常造血细胞,获得克隆优势。当前MDS研究面临两大关键问题:一是疾病机制尚未完全阐明——克隆优势与分化障碍的因果关系不明确;二是缺乏能忠实重现临床特征(如克隆竞争、多谱系发育异常、外周血减少、进展为急性白血病)的动物模型。现有基因工程小鼠模型(如Tet2-/-)虽能模拟发育异常,但难以重现“克隆竞争”;异种移植模型(如NOD/SCID小鼠)则因免疫排斥难以维持患者来源的克隆。
前期研究发现,孤儿核受体EAR-2(NR2F6)在MDS和急性髓系白血病(AML)患者的骨髓中高表达,且体外实验显示其能抑制白血病细胞系的分化、促进增殖。然而,EAR-2在体内造血系统中的功能尚不清楚,其是否通过调控分化诱导MDS样病变仍是未解决的问题。本研究旨在通过构建EAR-2过表达的小鼠骨髓嵌合体模型,探究EAR-2对体内造血分化的影响,填补“EAR-2体内功能”的研究空白,为MDS的机制研究提供新模型。
2. 文献综述解析
作者对MDS领域现有研究的评述逻辑围绕“疾病核心矛盾”与“模型缺陷”展开:首先,MDS的核心矛盾是“骨髓细胞凋亡增加但克隆优势”,现有研究提出“克隆干细胞的分化障碍”可能是关键——即恶性干细胞因分化受阻获得增殖优势,而分化的子代细胞因凋亡被清除,但这一假设缺乏体内实验验证;其次,现有MDS动物模型存在明显局限:基因工程小鼠模型多基于单一基因突变,难以模拟疾病异质性;异种移植模型则因人类造血干细胞难以在小鼠体内长期重建,无法模拟疾病进展。
针对EAR-2的研究,现有文献的关键结论包括:1)EAR-2在MDS/AML患者骨髓中高表达,其表达水平与细胞分化程度负相关;2)体外实验中,EAR-2过表达能抑制白血病细胞系(如32D)的分化,沉默则促进分化;3)EAR-2作为核受体,可能通过招募组蛋白去乙酰化酶(HDAC)发挥转录抑制作用。但现有研究的局限性在于:仅关注EAR-2的体外功能,缺乏体内造血系统的功能验证,更未探讨其与MDS发生的因果关系。
本研究的创新点在于:首次利用“逆转录病毒转导+骨髓嵌合体”模型,在体内验证EAR-2对造血分化的抑制作用,并证明其能诱导MDS的核心表型(克隆优势、发育异常、外周血减少、进展为白血病)。这一模型弥补了现有MDS模型的不足,为解析“分化障碍-克隆优势”机制提供了新工具。
3. 研究思路总结与详细解析
整体研究目标是探究EAR-2过表达对体内造血分化的影响,验证其是否能诱导MDS样病变;核心科学问题是“EAR-2是否通过抑制造血分化,在体内诱导MDS的核心表型”;技术路线为:体外逆转录病毒转导EAR-2至小鼠骨髓细胞→分选GFP+细胞并移植到致死照射受体→分析嵌合体的体内造血表型→体外实验验证分化与克隆能力→shRNA沉默EAR-2验证功能→机制研究(DNA结合域、HDAC依赖)。
3.1 骨髓细胞逆转录病毒转导与移植嵌合体构建
实验目的:获得EAR-2过表达的骨髓细胞,建立体内竞争模型,观察EAR-2对造血重建的影响。
方法细节:从C57BL/6小鼠获取骨髓细胞,用含EAR-2-IRES-GFP的MMP逆转录病毒转导(前期研究构建的载体),通过流式细胞术(FACS)分选GFP+细胞;将转导细胞(25%)与未转导细胞(75%)混合,移植到经致死照射的同系受体小鼠,构建造血嵌合体;对照组用空载体(GFP)转导。
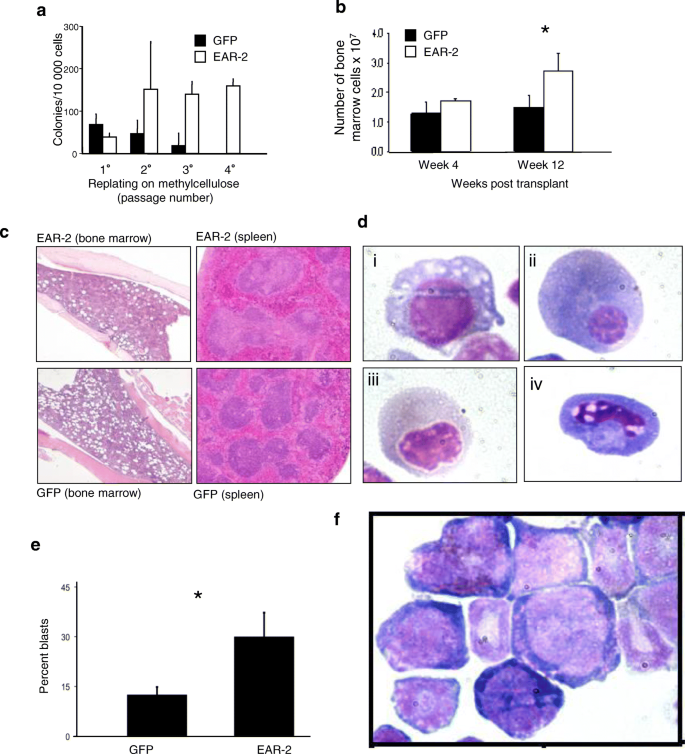
结果解读:EAR-2转导组的GFP+细胞在受体骨髓中的比例显著高于输入比例(附加文件1:图S1a),提示EAR-2过表达细胞具有竞争优势;移植后12周,EAR-2嵌合体的骨髓细胞密度显著高于对照组(图1b),且出现MDS特征:红细胞发育异常(图1d)、原始细胞比例增加(图1e)、未成熟前体细胞异常定位(ALIP,附加文件1:图S1b-c);二次移植后,受体全部发展为急性白血病(n=15),表现为骨髓、脾脏、肝脏的白血病细胞浸润(图2b)。

产品关联:实验所用关键产品包括Stem Cell Technologies的Methocult GF 3434甲基纤维素培养基(用于集落形成实验)、Becton Dickinson的LSR II流式细胞仪(用于细胞分选与表型分析);逆转录病毒载体(MMP)为前期研究构建,未提及具体商业品牌。
3.2 体内造血谱系分化分析
实验目的:验证EAR-2过表达对体内造血谱系分化的影响。
方法细节:对移植后5周和12周的嵌合体骨髓,用流式细胞术检测粒细胞(CD11b+GR-1+)、B细胞(B220+)等谱系的比例。
结果解读:EAR-2过表达组的粒细胞比例显著低于对照组(图3a-c),提示髓系分化受抑制;B220+细胞比例略有增加,但作者推测可能是EAR-2对B细胞分化的非特异性影响,未明确是否为功能性B细胞。

3.3 体外造血分化与克隆能力验证
实验目的:排除体内微环境影响,验证EAR-2对造血分化的细胞自主性作用。
方法细节:将EAR-2转导的骨髓细胞接种于甲基纤维素培养基(检测集落形成),或悬浮培养(含IL-3、IL-6、c-kit配体),用流式细胞术检测分化标志物(CD117:造血祖细胞;CD11b/GR-1:粒细胞);用shRNA沉默EAR-2(前期验证的shRNA,沉默效率~75%),检测集落大小与克隆能力。
结果解读:EAR-2过表达组的髓系集落(CFU-GM)和红系集落(BFU-E)数分别减少42.9%和34.6%(图4a),集落大小减少49.4%(附加文件2:图S4a);悬浮培养中,EAR-2过表达细胞保持CD117+表型,未表达CD11b/GR-1(图4b i-ii),提示分化受阻;serial replating实验显示,EAR-2过表达细胞的克隆能力显著强于对照组(能连续 replate多次,图4c)。沉默EAR-2后,集落大小显著增大(附加文件2:图S4c),且细胞分化为成熟粒细胞(CD11b+GR-1+,图4f),serial replating能力降低(图4d iv)。

3.4 EAR-2功能的机制研究
实验目的:探究EAR-2抑制分化的分子机制(DNA结合域与HDAC依赖)。
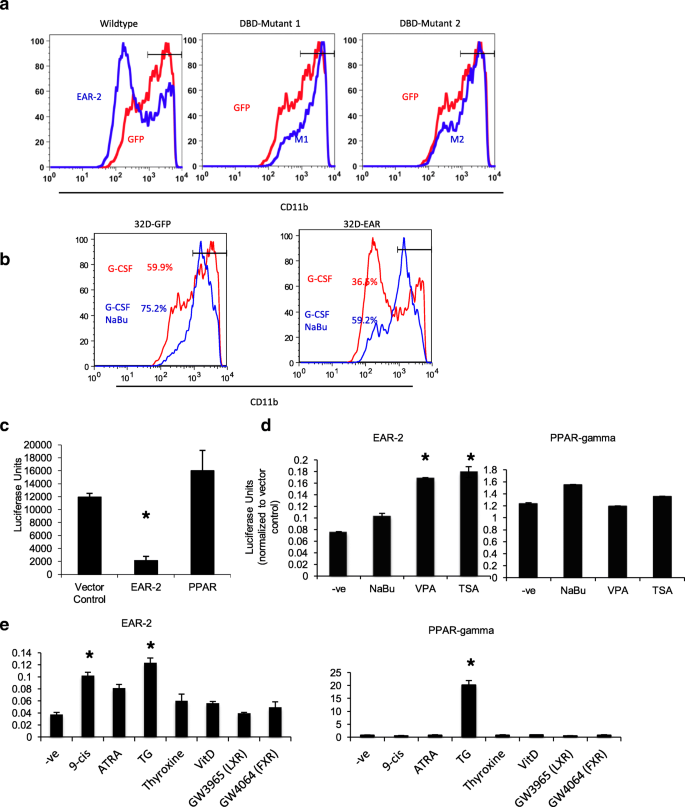
方法细节:构建EAR-2的DNA结合域突变体(P-box或D-box突变,破坏锌指结构),转导32D细胞(粒细胞系);用HDAC抑制剂(丁酸钠)处理EAR-2过表达的32D细胞,检测G-CSF诱导的分化(CD11b表达);构建Gal4-EAR-2融合蛋白,通过报告基因实验检测转录活性。
结果解读:DNA结合域突变体无法抑制32D细胞的分化(图5a),提示EAR-2的功能依赖DNA结合;丁酸钠处理恢复了EAR-2过表达细胞对G-CSF的响应,CD11b表达增加(图5b),提示HDAC参与抑制分化;报告基因实验显示,EAR-2融合蛋白能显著抑制 luciferase活性(图5c),且HDAC抑制剂(丙戊酸、曲古霉素A)能解除抑制(图5d),证明EAR-2作为转录抑制因子,依赖HDAC发挥作用。
4. Biomarker研究及发现成果解析
本研究中,EAR-2并非传统的“诊断/预后Biomarker”,而是作为“功能型Biomarker”——其异常表达(过表达)是MDS发生的驱动因素。筛选与验证逻辑为:前期研究从MDS/AML患者样本中筛选到EAR-2高表达→体外实验验证其抑制分化的功能→本研究通过体内模型验证其诱导MDS的表型。
研究过程详述:EAR-2的来源包括MDS/AML患者的骨髓样本(前期研究,n=12 MDS、n=15 AML)和小鼠骨髓细胞;验证方法包括qPCR(患者样本中EAR-2 mRNA高表达)、Western blot(shRNA沉默效率~75%)、功能实验(过表达诱导MDS表型,沉默促进分化);特异性数据:患者中EAR-2表达水平显著高于正常对照(前期研究,P<0.05),但本研究未提供ROC曲线等诊断性能数据。
核心成果提炼:EAR-2过表达能诱导MDS的核心表型,包括:1)造血干细胞竞争优势(GFP+细胞比例增加);2)多谱系发育异常(红细胞、粒细胞dysplasia);3)外周血减少(表1);4)进展为急性白血病(二次移植后100%发病,n=15)。其创新性在于首次证明EAR-2是MDS的驱动基因,为MDS的“分化障碍-克隆优势”机制提供了直接证据;统计学结果显示,EAR-2嵌合体的原始细胞比例(图1e)、粒细胞比例(图3a-c)与对照组有显著差异(P<0.05,文献未明确具体数值),二次移植的生存率显著低于对照组(图2a)。
本研究虽未将EAR-2开发为诊断标志物,但证明其异常表达是MDS发生的关键驱动因素,为MDS的靶向治疗提供了新靶点——抑制EAR-2的转录活性(如HDAC抑制剂)可能成为MDS的治疗策略。