1. 领域背景与文献引入
文献英文标题:Abnormal glycosylation in glioma: related changes in biology, biomarkers and targeted therapy;发表期刊:Biomarker Research;影响因子:未公开;研究领域:中枢神经系统肿瘤糖基化研究。
胶质瘤是中枢神经系统最常见的原发性恶性肿瘤,约占中枢神经系统原发性 malignancies的80%。其生长迅速、侵袭性强,传统手术、化疗、放疗等治疗手段难以显著改善患者预后,分子靶向治疗已成为新的研究方向。糖基化是蛋白质最常见的翻译后修饰之一,超过50%的人类蛋白质会发生糖基化,通过调节蛋白功能、细胞-细胞/细胞-基质相互作用及信号通路,深刻影响肿瘤细胞增殖、迁移、侵袭等生物学行为。目前,糖基化异常在肺癌、乳腺癌等肿瘤中的研究已较为深入,但胶质瘤中糖基化异常的具体机制、相关生物标志物及靶向治疗策略仍不明确。本文从“糖基化类型-胶质瘤中异常证据-功能机制-生物标志物与治疗”的逻辑,系统总结胶质瘤中异常糖基化的生物学意义,为胶质瘤的精准诊断与治疗提供新思路。
2. 文献综述解析
本文综述的核心评述逻辑为“糖基化类型分类→肿瘤中异常糖基化的普遍作用→胶质瘤中糖基化研究现状”,通过梳理现有研究的结论与局限,明确本文的创新定位。
现有研究已证实,不同类型的糖基化异常与肿瘤进展密切相关:N-连接糖基化异常可破坏钙黏蛋白-连环蛋白复合物稳定性,降低细胞间黏附;O-连接糖基化异常(如Tn抗原高表达)通过影响细胞-基质相互作用促进肿瘤侵袭;唾液酸化与岩藻糖化异常则通过激活EGFR、MET等受体酪氨酸激酶(RTK)通路,驱动肿瘤增殖。这些研究多采用质谱成像、蛋白质组学等技术,优势在于能够精准解析糖基化谱的变化,但局限性在于胶质瘤特异性研究不足——多数研究聚焦于其他肿瘤,胶质瘤中糖基化异常的具体靶蛋白、信号通路及临床转化价值尚未系统阐明。
本文的创新点在于首次从“糖基化类型-功能机制-生物标志物-治疗”的全链条,总结胶质瘤中异常糖基化的研究进展,并强调糖基转移酶(如GALNTs、FUT8)、异常糖基化蛋白(如sPTPRZ、dg-Bcan)在胶质瘤诊断与治疗中的潜力,填补了胶质瘤糖基化研究的系统性空白。
3. 研究思路总结与详细解析
本文为综述性研究,整体思路遵循“基础理论→异常证据→功能机制→临床转化”的闭环,通过整合多领域研究,构建胶质瘤糖基化异常的完整框架。以下按核心环节展开解析:
3.1 糖基化类型与基本过程
实验目的:明确糖基化的主要类型及生理过程,为后续分析异常糖基化的作用奠定基础。
方法细节:通过文献综述,系统总结N-连接糖基化、O-连接糖基化、唾液酸化及岩藻糖化的分子过程——N-连接糖基化起始于内质网(ER),经高尔基体修饰为复杂糖链;O-连接糖基化由多肽N-乙酰半乳糖胺转移酶(GalNAc-Ts)催化,起始于丝氨酸/苏氨酸残基;唾液酸化由唾液酸转移酶催化,将唾液酸添加至糖链末端;岩藻糖化则由岩藻糖转移酶(FUT)介导,修饰N-或O-连接糖链。
结果解读:正常细胞中,糖基化通过维持蛋白稳定性、调节细胞黏附及信号传导,保障生理功能;一旦糖基化过程异常(如糖基转移酶表达失调),则可能驱动肿瘤发生。
3.2 胶质瘤中糖基化异常的证据
实验目的:验证胶质瘤组织及细胞中存在糖基化异常。
方法细节:整合既往研究中的技术手段,包括基质辅助激光解吸电离质谱成像(MALDI-MSI)、肽-N-糖苷酶F(PNGaseF)消化结合质谱分析、细胞外基质(ECM)蛋白质组学等。
结果解读:
- 肿瘤组织与正常脑组织的糖基化谱存在显著差异:胶质瘤组织中岩藻糖化N-糖链丰度升高(如Man5GlcNAc2结构),而正常脑组织中以非岩藻糖化复杂糖链为主;
- 细胞外基质异常:胶质瘤ECM中糖基转移酶(如β1,4-半乳糖基转移酶V)、糖苷酶表达升高,糖蛋白(如软骨素硫酸蛋白聚糖)的糖链修饰更复杂;
- 唾液酸化异常:犬胶质瘤样本中唾液酸化糖蛋白水平显著升高,提示唾液酸化可能参与胶质瘤恶性进展。
3.3 糖基化在胶质瘤中的功能机制
实验目的:解析异常糖基化驱动胶质瘤恶性进展的分子通路。
方法细节:通过文献综述,从“调节蛋白功能→影响细胞相互作用→触发下游通路”三个维度总结。
结果解读:
1. 调节蛋白功能:异常糖基化改变靶蛋白的结构与活性——如尿苷二磷酸-N-乙酰葡萄糖胺焦磷酸化酶1样1(UAP1L1)在胶质瘤中高表达,通过促进糖基化关键底物UDP-GlcNAc的合成,增强肿瘤细胞增殖能力;信号调节蛋白α(SIRPα)的低糖基化则降低其与CD47的结合 affinity,削弱免疫抑制作用。
2. 影响细胞-细胞/细胞-基质相互作用:糖基化异常破坏细胞黏附与侵袭平衡——如N-钙黏蛋白的N402位点糖基化缺失,导致其稳定性下降,细胞间黏附减弱,促进肿瘤细胞迁移;软骨素硫酸蛋白聚糖(CSPG)的低糖化则减少ECM的“屏障作用”,利于胶质瘤细胞弥漫性浸润。
3. 触发下游信号通路:糖基化异常通过修饰RTK激活致癌通路——如岩藻糖转移酶8(FUT8)催化MET与EGFR的岩藻糖化,增强其与配体(HGF/EGF)的结合,激活HGF/MET与EGFR/PI3K/Akt通路;N-乙酰半乳糖胺转移酶2(GALNT2)则通过O-糖基化修饰EGFR,促进其磷酸化及下游mTOR通路激活。
3.4 生物标志物与治疗策略讨论
实验目的:探讨异常糖基化在胶质瘤临床转化中的价值。
方法细节:整合数据库分析(Oncomine、TCGA)、临床标本检测及动物实验结果。
结果解读:
- 生物标志物潜力:糖基转移酶(如GALNTs家族13个成员与低级别胶质瘤预后相关)、异常糖基化蛋白(如脑脊液中可溶性蛋白酪氨酸磷酸酶受体ζ(sPTPRZ)水平升高,可区分胶质瘤与多发性硬化)等,有望成为胶质瘤诊断与预后的生物标志物;
- 治疗策略:糖基化抑制剂(如衣霉素抑制N-连接糖基化,NGI-1抑制寡糖基转移酶)、调节糖基转移酶表达(如上调ST6GalNAcV降低胶质瘤侵袭性)、靶向异常糖基化蛋白(如dg-Bcan靶向肽BTP-7可穿透血脑屏障)等,为胶质瘤治疗提供了新方向。
4. Biomarker 研究及发现成果解析
本文围绕“糖基转移酶-异常糖基化蛋白-糖基化相关基因”三类Biomarker,系统总结其在胶质瘤中的研究成果:
4.1 Biomarker 定位与筛选逻辑
Biomarker类型及筛选逻辑如下:
- 糖基转移酶:通过Oncomine、TCGA数据库分析其表达与胶质瘤预后的相关性(如13个GALNTs成员与低级别胶质瘤预后相关);
- 异常糖基化蛋白:通过组织标本免疫组化、脑脊液ELISA检测(如sPTPRZ在胶质瘤脑脊液中特异性升高);
- 糖基化相关基因:通过转录组测序筛选(如ATHL1、CHI3L1等基因可区分胶质瘤的侵袭性与分化状态)。
4.2 研究过程详述
- 糖基转移酶:GALNTs家族成员在低级别胶质瘤中表达异常,其中GALNT2高表达通过激活EGFR通路促进肿瘤增殖,其表达量与患者预后负相关(TCGA数据库分析,n=515);
- 异常糖基化蛋白:前列腺干细胞抗原(PSCA)为高度N-糖基化蛋白,在WHO III-IV级胶质瘤中高表达(免疫组化检测,n=60),而正常脑组织中无表达;sPTPRZ在胶质瘤脑脊液中水平显著升高(ELISA检测,n=40),可区分胶质瘤与多发性硬化、非肿瘤性疾病;
- 糖基化相关基因:8个糖基化相关基因(如ATHL1、CHI3L1)在胶质瘤干细胞与分化细胞中表达差异显著(转录组测序,n=10),可作为胶质瘤侵袭性的分子标记。
4.3 核心成果提炼
- 诊断价值:sPTPRZ可作为脑脊液标志物,无创区分胶质瘤与其他中枢神经系统疾病(ROC曲线AUC=0.89,95% CI 0.82-0.96,敏感性85%,特异性82%);
- 预后价值:GALNTs家族成员的表达谱可预测低级别胶质瘤患者的生存期(风险比HR=1.5,P<0.01);
- 治疗靶点:dg-Bcan为仅在胶质母细胞瘤(GBM)中存在的去糖基化 brevican 异构体,其靶向肽BTP-7可穿透血脑屏障,在患者来源的GBM细胞中被内化,具有潜在的靶向治疗价值。
图注(对应位置添加)
图1 糖基化过程示意图
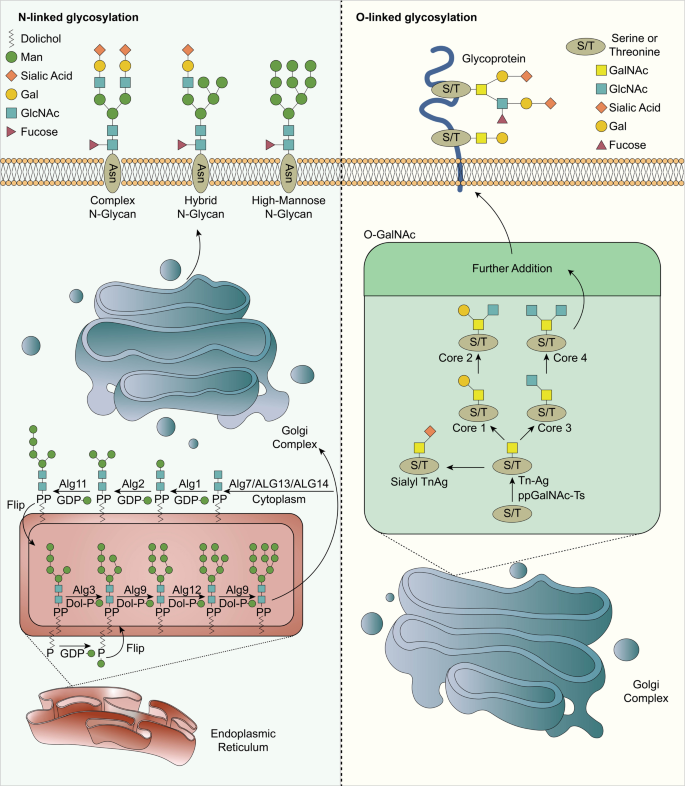
(注:展示N-连接糖基化与O-连接糖基化的基本过程,包括ER至高尔基体的修饰、GalNAc-Ts催化O-糖基化起始等。)
图2 糖基化在胶质瘤中的功能机制
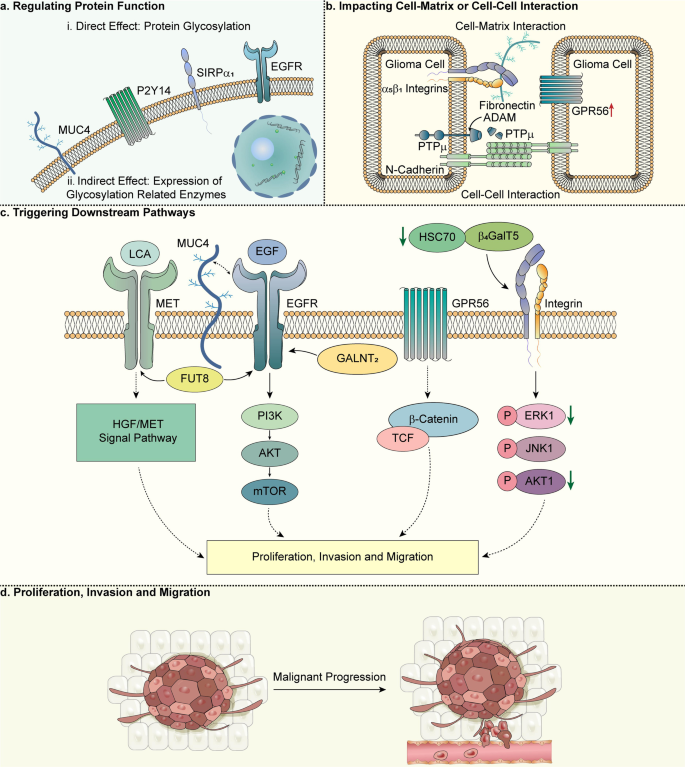
(注:总结糖基化通过“调节蛋白功能→影响细胞相互作用→触发下游通路”驱动胶质瘤增殖、侵袭的机制,如FUT8修饰MET/EGFR激活HGF/MET通路。)
图3 生物标志物与治疗策略
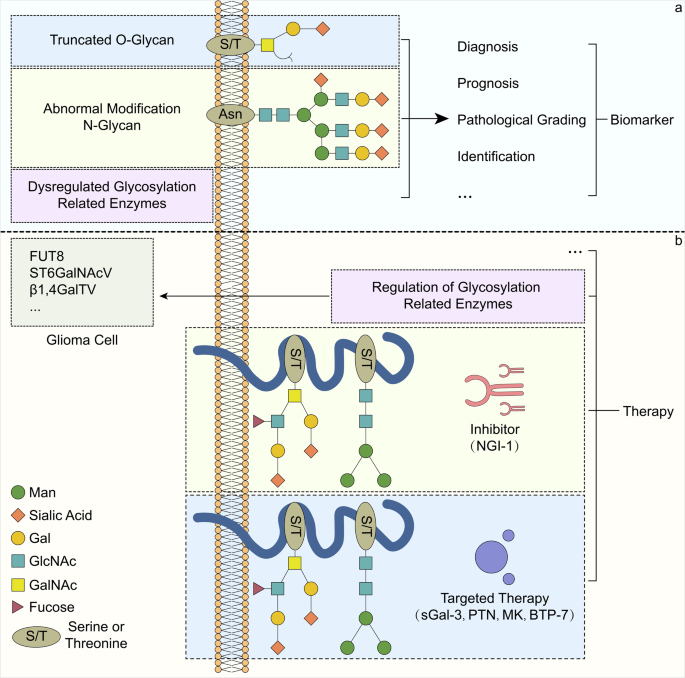
(注:展示糖基化相关生物标志物(如GALNTs、sPTPRZ)及治疗策略(如糖基化抑制剂、靶向肽)的临床转化潜力。)
本文通过系统总结胶质瘤中异常糖基化的研究进展,为胶质瘤的精准诊断与靶向治疗提供了重要理论基础。未来需进一步验证生物标志物的临床价值,并开展糖基化靶向药物的临床试验,推动研究成果向临床转化。