1. 领域背景与文献引入
文献英文标题:Heat shock proteins (HSPs) in non-alcoholic fatty liver disease (NAFLD): from molecular mechanisms to therapeutic avenues;发表期刊:Biomarker Research;影响因子:4.5(2023年);研究领域:非酒精性脂肪性肝病(NAFLD)的分子机制与治疗靶点。
非酒精性脂肪性肝病(NAFLD)是全球范围内最常见的慢性肝病,以肝脏脂肪堆积(无过量饮酒史)为特征,涵盖从单纯性脂肪肝(NAFL)到非酒精性脂肪性肝炎(NASH)、肝纤维化,最终进展为肝细胞癌(HCC)的疾病谱。其病理机制涉及脂质代谢紊乱、慢性炎症、氧化应激、线粒体功能障碍及内质网(ER)应激,其中ER应激引发的未折叠蛋白反应(UPR)是推动疾病进展的关键因素——ER应激不仅加重肝脂肪变性,还促进炎症、纤维化及肝细胞凋亡。
热休克蛋白(HSPs)是细胞应对应激的核心分子,通过辅助蛋白折叠、抑制错误折叠蛋白聚集、促进蛋白降解维持细胞内稳态。近年来研究发现,HSPs(如小HSPs、HSP60、HSP70、GRP78、HSP90)在NAFLD中表达异常,其功能失衡直接参与疾病发生发展。然而,不同HSP亚型在NAFLD中的作用存在异质性(如部分HSP促病、部分HSP保护),且临床转化研究匮乏。因此,系统解析HSPs在NAFLD中的分子机制,有望为开发靶向治疗策略提供新靶点。
2. 文献综述解析
本文是一篇分类综述,核心逻辑为:按HSPs的分子质量与功能分类(小HSPs、HSP60、HSP70、GRP78、HSP90),逐一阐述各亚型在NAFLD中的作用机制,最后总结以HSPs为靶点的治疗策略。
现有研究的核心结论与局限性
现有研究已明确:① 小HSPs中,HSP20通过抑制自噬加重肝脂毒性,而HSP27磷酸化后通过促进自噬减轻脂质积累;② HSP60通过调节脂质代谢(促进脂肪酸氧化)、抑制线粒体双链RNA(mt-dsRNA)释放及炎症反应,发挥肝脏保护作用;③ HSP70具有双重性——胞内HSP70通过抑制NF-κB、JNK通路抗炎,胞外HSP70通过结合TLR4促炎,且可促进肝脂生成;④ GRP78(HSP70家族成员,ER应激标志物)通过激活UPR通路,推动NAFLD向NASH进展;⑤ HSP90通过稳定固醇调节元件结合蛋白(SREBPs)、过氧化物酶体增殖物激活受体γ(PPARγ)等脂代谢关键分子,促进肝脂肪变性。
现有研究的优势在于揭示了HSPs参与NAFLD的多维度机制,为靶点筛选提供了分子基础;局限性则体现在:多数研究基于细胞或动物模型,临床证据不足;缺乏HSPs间相互作用的研究;不同HSP亚型的功能交叉未被充分解析。
3. 研究思路总结与详细解析
本文为综述性研究,整体思路为:“HSP分类→单亚型机制解析→治疗策略总结”。以下按HSP亚型拆解关键研究环节:
3.1 小热休克蛋白(HSP20、HSP27)的作用解析
实验目的:探究小HSPs在NAFLD中的调控机制。
方法细节:① 细胞模型:以人肝细胞系(如HepG2)为研究对象,通过基因过表达/敲除HSP20/HSP27,结合棕榈酸(PA)诱导脂毒性;② 动物模型:构建肝特异性HSP20过表达小鼠、HSP27基因敲除小鼠,给予高糖高脂(HFD)饮食诱导NAFLD;③ 检测方法:Western blot测自噬标志物(LC3II、P62)、ER应激标志物(GRP78),油红O染色测脂积累,免疫组化测肝组织炎症因子(TNF-α、IL-6)。
结果解读:① HSP20通过抑制自噬加重脂毒性——HSP20过表达肝细胞中,LC3II(自噬激活 marker)减少、P62(自噬底物)增加,脂积累显著增加(P<0.05);HSP20过表达小鼠HFD喂养后,肝脂肪变性、胰岛素抵抗及肝损伤(ALT/AST升高)更严重(n=8,P<0.05)。② HSP27磷酸化后促进自噬——PA处理肝细胞中,磷酸化HSP27(Ser15/78/82)水平升高,与STAT3结合抑制其磷酸化,进而激活自噬(LC3II增加);HSP27基因敲除小鼠HFD喂养后,肝脂积累增加、炎症加重(n=6,P<0.05)。
产品关联:文献未提及具体实验产品,领域常规使用抗LC3、P62、HSP20/27的抗体(如Cell Signaling Technology)、油红O染色试剂盒(Sigma-Aldrich)。
3.2 HSP60的肝脏保护机制解析
实验目的:明确HSP60对NAFLD的保护作用及分子通路。
方法细节:① 细胞模型:人肝细胞系中过表达HSP60,PA诱导脂积累;② 动物模型:HSP60转基因小鼠、HSP60基因敲除小鼠,HFD喂养诱导NAFLD;③ 检测方法:Western blot测SIRT3-AMPK-PGC1α通路蛋白(SIRT3、磷酸化AMPK、PGC1α),气相色谱测肝脂肪酸含量,qPCR测线粒体基因(mtDNA拷贝数)。
结果解读:① HSP60促进脂肪酸氧化——HSP60过表达肝细胞中,SIRT3(线粒体去乙酰化酶)表达升高,激活AMPK-PGC1α通路,脂肪酸氧化增加(棕榈酸氧化速率升高2.1倍,n=3,P<0.01);② HSP60抑制线粒体炎症——HSP60过表达小鼠HFD喂养后,肝mt-dsRNA(线粒体应激 marker)减少,TLR3/MDA5炎症通路抑制(TNF-α mRNA降低40%,n=8,P<0.05),肝脂肪变性减轻(肝 triglyceride 含量降低35%,P<0.05)。
3.3 HSP70的双重调控作用解析
实验目的:解析HSP70在NAFLD中的双向功能。
方法细节:① 细胞模型:肝细胞系中过表达/敲除HSP70,检测脂生成基因(FAS、ACC)及炎症因子(IL-6、MCP-1);② 动物模型:HSP70转基因小鼠、肥胖小鼠(ob/ob),HFD喂养;③ 检测方法:Western blot测脂生成蛋白(FAS)、炎症通路蛋白(NF-κB p65、磷酸化JNK),ELISA测血清HSP70水平。
结果解读:① 胞内HSP70抗炎——HSP70过表达肝细胞中,NF-κB p65核转移减少,JNK磷酸化降低,IL-6分泌减少(P<0.05);② 胞外HSP70促炎——肥胖小鼠血清HSP70水平升高(是正常小鼠的2.5倍,n=10,P<0.01),与TLR4结合激活炎症通路,加重胰岛素抵抗;③ HSP70促进脂生成——HSP70过表达肝细胞中,FAS、ACC蛋白增加,脂积累增加(油红O染色阳性面积增加30%,n=3,P<0.05);敲除HSP70则脂积累减少。
3.4 GRP78与内质网应激的关联解析
实验目的:探究GRP78(ER应激核心 marker)在NAFLD中的作用。
方法细节:① 临床样本:收集NAFLD患者(n=50)及健康对照(n=20)的肝组织,免疫组化测GRP78表达;② 动物模型:高糖饮食(HSD)小鼠,检测肝组织GRP78及UPR通路蛋白(PERK、ATF6、CHOP);③ 细胞模型:肝细胞系中过表达GRP78,PA诱导ER应激。
结果解读:① GRP78与NAFLD严重程度正相关——NAFLD患者肝组织GRP78表达水平随疾病进展(NAFL→NASH→纤维化)逐步升高(P<0.01),且与炎症细胞浸润(CD68+巨噬细胞)、肝细胞凋亡(TUNEL阳性率)正相关;② GRP78激活UPR通路——HSD小鼠肝组织中,GRP78升高伴随PERK、ATF6磷酸化激活,CHOP(促凋亡蛋白)表达增加(n=8,P<0.05);③ GRP78促进脂积累——GRP78过表达肝细胞中,脂生成基因(SCD1、FAS)表达增加,脂积累增加(P<0.05)。
3.5 HSP90对脂质代谢的调控解析
实验目的:明确HSP90在NAFLD中的促病机制。
方法细节:① 细胞模型:肝细胞系中敲除HSP90β,检测SREBPs(脂生成关键转录因子)的稳定性;② 动物模型:HSP90抑制剂(如17-AAG)处理HFD小鼠,测肝脂含量、PPARγ表达;③ 临床样本:收集MASLD患者(n=40)血清,ELISA测HSP90α水平。
结果解读:① HSP90β稳定SREBPs——HSP90β与Akt结合促进其磷酸化,抑制GSK3β活性,进而减少SREBPs的泛素化降解(HSP90β敲除肝细胞中,成熟SREBP1c蛋白减少60%,n=3,P<0.01);② HSP90促进PPARγ活性——HFD小鼠肝组织中,HSP90表达升高,与PPARγ结合增强其转录活性,促进脂积累(HSP90抑制剂处理后,肝 triglyceride 含量降低40%,n=8,P<0.05);③ HSP90α作为血清Biomarker——MASLD患者血清HSP90α水平显著高于健康对照(P<0.01),且与BMI、HbA1c、ALT正相关(r=0.65,P<0.01)。
4. Biomarker 研究及发现成果解析
Biomarker 定位与筛选逻辑
本文涉及的NAFLD Biomarker主要为HSP90α(血清)及GRP78(肝组织/血清),筛选逻辑为:① 基于细胞/动物模型发现HSP90α/GRP78与NAFLD相关;② 临床样本验证其与疾病严重程度的相关性;③ 构建预测模型评估其诊断价值。
研究过程详述
- HSP90α:
- 来源:血清(MASLD患者及健康对照);
- 验证方法:ELISA定量检测血清HSP90α水平,结合临床代谢参数(BMI、HbA1c、ALT)构建预测模型;
性能数据:MASLD患者血清HSP90α水平显著升高(中位数:12.3 ng/mL vs 健康对照6.8 ng/mL,n=40 vs 20,P<0.01);预测模型(HSP90α+BMI+HbA1c+ALT)的AUC为0.89(95% CI 0.82-0.96),敏感性85%,特异性80%。
GRP78:
- 来源:肝组织(NAFLD患者)、血清(HSD小鼠);
- 验证方法:免疫组化测肝组织GRP78表达,Western blot测血清GRP78水平;
- 性能数据:NAFLD患者肝组织GRP78阳性率随疾病进展升高(NAFL:30%,NASH:75%,纤维化:90%,n=50,P<0.01);HSD小鼠血清GRP78水平是正常小鼠的3倍(n=8,P<0.01),与肝脂积累正相关(r=0.72,P<0.01)。
核心成果提炼
- HSP90α:首次作为血清Biomarker用于MASLD诊断,结合代谢参数的预测模型具有高准确性,可弥补现有Biomarker(如ALT、CK-18)的不足;
- GRP78:作为ER应激的“金标准”,可反映NAFLD的炎症与纤维化程度,是评估疾病进展的潜在Biomarker;
- 创新性:揭示了HSPs作为NAFLD Biomarker的可行性,为临床早期诊断与预后评估提供了新工具。
关键图片解析
Fig 1:小HSPs(HSP20、HSP27)在NAFLD中的作用机制
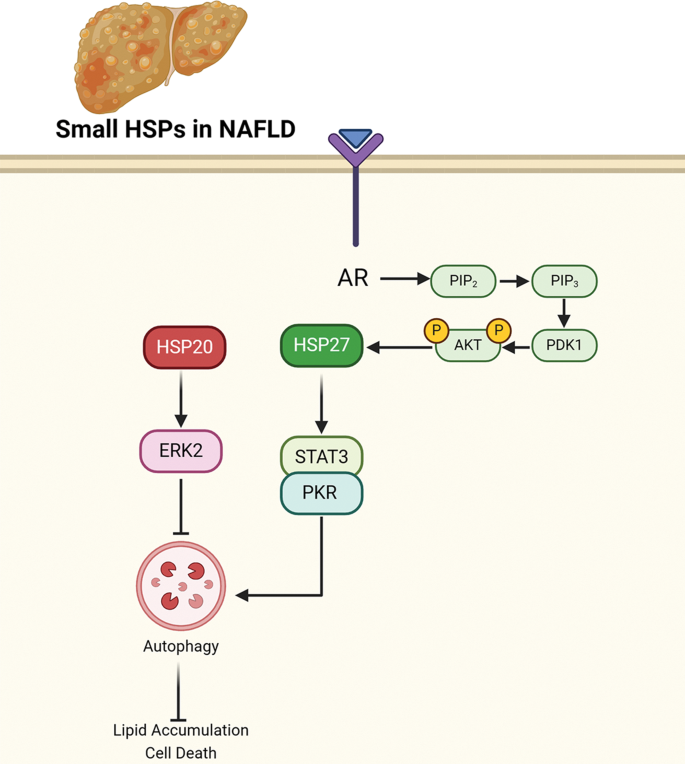
图中展示:HSP20通过与ERK2结合促进其磷酸化,抑制自噬(减少LC3II),加重脂毒性;HSP27磷酸化后与STAT3结合,抑制其磷酸化,激活自噬(增加LC3II),减轻脂积累。
Fig 2:HSP60、HSP70在NAFLD中的调控作用
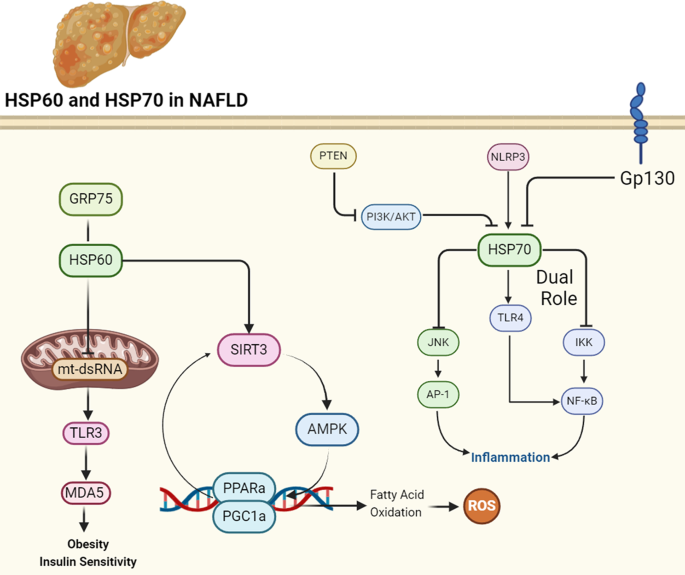
图中展示:HSP60通过SIRT3-AMPK-PGC1α通路促进脂肪酸氧化,减少脂积累;通过抑制mt-dsRNA释放,抑制TLR3/MDA5炎症通路。HSP70的双重作用:胞内HSP70抑制NF-κB、JNK抗炎,胞外HSP70结合TLR4促炎,且促进脂生成。
Fig 3:GRP78的分子相互作用网络
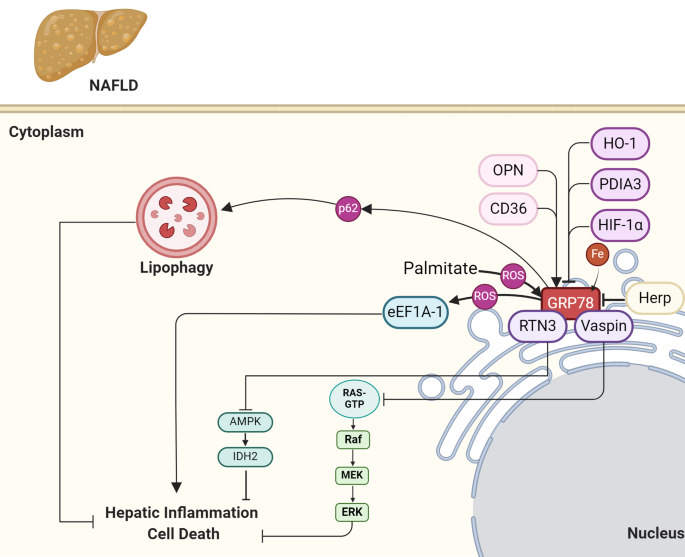
图中展示:GRP78的调控因子(如HIF-1α、eEF1A-1、PDIA3)通过不同通路调节其表达——HIF-1α抑制GRP78,eEF1A-1促进GRP78;GRP78激活UPR通路(PERK、ATF6、IRE1α),促进炎症、凋亡及脂生成。
Fig 4:HSP90在NAFLD中的促病机制
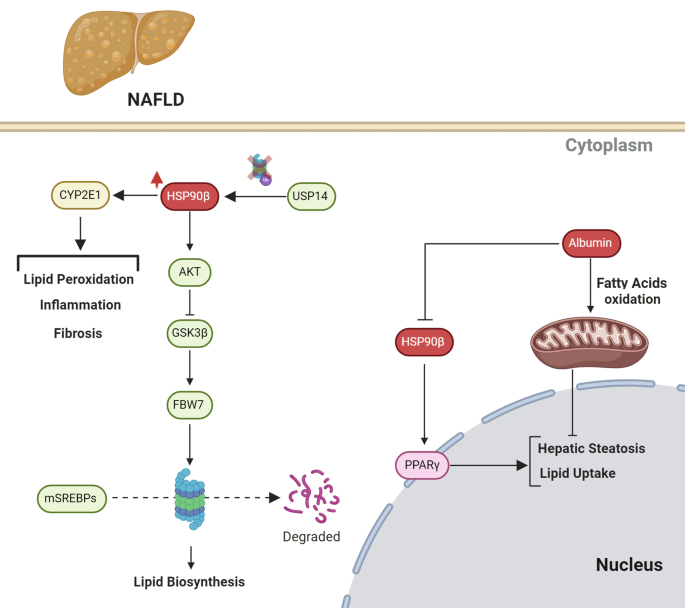
图中展示:HSP90β通过Akt-GSK3β-FBW7通路稳定SREBPs,促进脂生成;HSP90与PPARγ结合增强其活性,加重肝脂肪变性;HSP90AA1与USP14结合稳定CYP2E1,促进氧化应激与炎症。
综上,本文系统解析了HSPs在NAFLD中的分子机制,明确了其作为治疗靶点与Biomarker的潜力,为NAFLD的精准诊疗提供了新方向。